
Evaluation of the dynamic electrochemical stability of ionic liquid–metal interfaces against reactive oxygen species using an in situ electrochemical quartz crystal microbalance†
Chunhui Xiao‡,
Abdul Rehman and
Xiangqun Zeng*
Department of Chemistry, Oakland University, Rochester, Michigan 48309, USA. E-mail: zeng@oakland.edu
First published on 12th March 2015
Abstract
The dynamic interactions between the electrochemically generated superoxide radical (O2˙−) and three structurally different ionic liquids (ILs) were characterized using an electrochemical quartz crystal microbalance (EQCM). We established the long-term stability of the interface (on the scale of hours and weeks) against the most ubiquitous interferent in electrochemical energy devices and sensors: oxygen. Oxygen potentially limits the application of IL electrolytes in these devices. In particular, the electrochemical behavior of the O2/O2˙− couple and the ion pair formed between the cation of an IL and O2˙− were evaluated. O2˙− tends to form ion pair complexes with the cation of an IL, subsequently abstracting a proton to form different products depending on the cationic structure of the IL used. The reversibility of the O2/O2˙− electrode reaction depends on the subsequent chemical reactions between O2˙− and the IL, which are more pronounced at slow scan rates. It was found that O2˙− was significantly more stable in the IL with the [BMPY] cation than in ILs with imidazolium salts. The stability of the ILs towards O2˙− attack follows the order [BMPY][NTf2] > [BdMIM][NTf2] > [BMIM][NTf2] as evaluated on a time-scale of a few seconds to minutes and up to 3 weeks. It was found that the formation of the [Cation]⋯O2˙− ion pair complex lowers the local viscosity of the IL near the electrode, reflected in the change in the oscillating frequency of the quartz crystal electrode. As much as is the feasibility for the formation of this ion pair, that much is the tendency of the IL to lose its electrochemical stability. A combined analysis can provide a quick indication of the dynamic stability of the electrode–electrolyte interface for any IL in the presence of certain electrochemical reactions using the EQCM technique.
Introduction
The electrode–electrolyte interface has a central role in electrochemical devices, including those devices that involve energy production and storage and those that are used as sensors. Ionic liquids (ILs)1 provide a range of options to diversify this interface as a result of their structural heterogeneity and synthetic flexibility. As a consequence, they have attracted much interest2–7 in recent years, especially in the demanding energy sectors of dye-sensitized fuel cells,6,8 batteries9–11 and supercapacitors,12–14 as well as in fundamental electrochemistry.15–18 However, it is very important to understand and quantify the stability of this interface in the presence of species that are ubiquitous under ambient conditions. Oxygen is one of these species, presence of which in many of these electrochemical reactions can generate reactive oxygen species. The superoxide radical (O2˙−) affects subsequent reactions and the stability of the interface. Therefore the thermodynamics, kinetics and even the mechanisms of the electrode reactions are changed, compromising the efficiency of the overall process. For example, recent work19 has shown that the mechanism of CO2 reduction at IL interfaces is very different in the presence and absence of oxygen. The study further showed that modification of the reaction mechanism occurs because of the competition between CO2 and the cations of the ILs reacting with the electrochemically generated O2˙− radical. This modification was shown to be dependent on the structure of the IL, indicating that oxygen affects the interface of different ILs to varying degrees. A dynamic measurement of subtle variations at the electrode–IL interface is therefore critical in ensuring that all these applications work, which, in turn, requires a more sophisticated methodology to quantify the effects than simple electrochemical techniques.Unlike in conventional organic solvents, the redox peaks of the O2/O2˙− couple in ILs are asymmetrical in shape, with the characteristics of a typical steady-state condition. This is a result of the specific interactions of O2˙− with the cation of the ILs.20–22 As superoxide has a negative charge and the electronic spin density is delocalized between the two oxygen atoms, it can act as a powerful nucleophile in aprotic solvents.23–25 This property has also been observed in ILs. For example, the superoxide generated in situ in ILs based on the imidazolium cation tends to attack positively charged organic cations, leading to strong ion pairing of the O2˙−⋯[Cation]+.26,27 The ion pairing phenomenon during the dioxygen reduction reaction in EMI+-based ILs has been thoroughly investigated.28 It was observed that, in DMSO solution, the degree of association of O2˙− with the EMI+ cation is comparable with that of O2˙− with H2O. However, as the degree of association in pure ILs is much stronger, such an ion pairing may ultimately lead to the abstraction of a proton from the cation to form HO2˙− and carbine, or other types of compounds,29–31 thus hampering the stability of the O2˙− species in the ILs. The cation structure in an IL plays an important part in this regard. Islam et al.32 reported that O2˙− can attack the 2-position of the imidazolium ring to form an ion pair complex. Eventually, the ion pair complex of the imidazolium cation and the O2˙− species undergoes a ring-opening reaction to form hydrogen peroxide. AlNashef et al.33 reported that O2˙− can react with alkyl imidazolium cations to yield 2-imidazolones. Hayyan et al.22 studied the generation of O2˙− in pyridinium-, morpholinium-, ammonium- and sulfonium-based ILs by cyclic voltammetry (CV) and chronoamperometry. They found that O2˙− is not stable in the pyridinium-based ILs, but is stable in the other ILs for the duration of the experiments. However, most experiments were performed to observe the stability of the superoxide at very short time-scales (from seconds to minutes) depending on the methods and techniques used. However, none of these studies estimated the stability of the ILs involved at the longer time-scales that are important in the real-world applications of these systems. In many of these studies, simulation models were used to fit the experimental parameters to obtain mechanistic information.
In contrast, we and others have shown19,34,35 that the use of an in situ electrochemical quartz crystal microbalance (EQCM) provides a better and powerful method to characterize the structure and dynamics of the properties of the IL–electrode interface, allowing real-time monitoring of variations in the surface populations during electrochemically driven redox reactions. The electrified interface causes the IL ions to rearrange, thereby generating different kinds of solvent polarization effects depending on the IL used, which determines the reaction rate. The high viscosity and resulting slow relaxation dynamics of ILs36 allow the time-scale of these polarization effects and the electron transfer processes to be at similar levels, thus allowing the measurement of subtle variations in the structure of the IL solvent during the redox reaction. The results can then be used to quantify the ionic interactions via adsorption on the electrode surface, ion pairing within the IL or with the reaction products, and the ionic arrangements. The data generated in this way (Scheme 1) enable us to account for different types of structural orientations under potential control during the electrochemical perturbation of the IL–electrode interface. Therefore it is thought that this approach may give an insight into various fundamental aspects of IL stability against certain species such as the O2˙− radical and the dynamics of redox reactions in ILs.
 | ||
| Scheme 1 Flow chart of the dynamic variables influenced by the structural heterogeneities in ILs as well as by their reactions with electrochemically generated species. These variables affect each other during electrochemical perturbations and can be detected by the EQCM system. | ||
In this work, the dynamic interactions between the electrochemically generated superoxide radical (O2˙−) and ILs with different structures were characterized by the EQCM approach to quantitatively estimate their stability against this radical. Specifically, we studied the interactions between the electro-generated superoxide and the ILs with two new focuses. First, we were motivated to characterize the reactivity of the in situ generated superoxide anion radicals in ILs at much longer time-scales (seconds to hours) by multiple cycles of CV and at various scan rates to quantitatively determine the stability of ILs in the presence of superoxide anion radicals. Second, we intended to demonstrate that the EQCM technique can measure the dynamics of the potential-induced subtle variations at an IL–electrode interface in the absence and presence of oxygen redox processes. The key emphasis is to understand how the IL structural heterogeneity of the interface contributes to the reaction kinetics by selectively interacting with the superoxide radical. Cumulatively, these analyses can provide new quantitative information regarding the oxygen reduction reaction (ORR) in ILs. This is important for the selection and design of new and stable electrolytes and for the utilization of O2˙− in a wide range of applications, such as developing highly selective electrochemical sensors and electrochemical energy storage devices using ILs.
Experimental
Chemicals
Hydrophobic 1-butyl-3-methylimidazolium bis(trifluoromethanesulfonyl)imide ([BMIM][NTf2]), 1-butyl-2,3-dimethylimidazolium bis(trifluoromethanesulfonyl)imide ([BdMIM][NTf2]) and 1-butyl-1-methylpyrrolidinium bis(trifluoromethanesulfonyl)imide ([BMPY][NTf2]) with purities of 99% were purchased from Ionic Liquids Technologies (USA). All the ILs were dried before use in the electrochemical experiments by heating to 120 °C in a vacuum oven for a minimum of 12 h until no visible signs of water were present in the IR spectrum (CaF2 plates). All other chemicals were purchased from Sigma-Aldrich (St. Louis, MO, USA) and were of analytical-reagent grade and used without further purification.Electrochemical measurements
Volumes of 100 μL of the respective ILs were drop-coated onto the working electrode inside a Kel-F electrochemical cell designed to facilitate the correct positioning of the three-electrode system. An Au quartz crystal (AT-cut 10 MHz, unpolished with an approximately 1000 Å thick Au film with an area of 0.23 cm2; International Crystal) was used as the working electrode. Just before use, the Au substrates were cleaned by a freshly prepared “piranha” solution for about 30 s and subsequently rinsed with excess amounts of water, ethanol and acetone, then dried under nitrogen gas (caution: the “piranha” solution, 1![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) :3 H2O2:H2SO4, reacts violently with many organic materials and should be used with extreme care; it should not be stored in a sealed container). A platinum wire and a silver wire (0.5 mm diameter) were used as the counter electrode and the quasi-reference electrode, respectively. They were mechanically polished prior to use to remove the oxide film, rinsed with acetone and then dried under vacuum. The distance from the tip of the two electrodes to the center of the working electrode was set at 2 mm to minimize the effects of uncompensated electrolyte resistance. All potentials were measured using Fc/Fc+ (50 μM ferrocene in ILs) as the internal reference. Prior to the measurements, the entire system was dried in a vacuum oven at 60 °C for at least 2 h to remove any water and other impurities. The total gas flow was fixed at 200 sccm by digital mass flow controllers (MKS Instruments). Two mass flow controllers were used to adjust the volume ratio of the test gas flows. The highly purified gases (N2 and O2) were used containing <10 ppm water as described by the manufacturers. All the gas analytes were prepared by pre-mixing the gases in a 500 mL glass bottle with a stirring fan and then connecting to a drying column consisting of silica gel and solid calcium chloride before their introduction into the electrochemical cell. Using this drying process, the water level could be limited to <10 ppm. The gas was run for at least 12 h to ensure that equilibrium was established before the measurements were made. The IL system was purged constantly during the experiments with dry nitrogen or a mixture of nitrogen and oxygen to prevent any trace amounts of water or other impurities. All experiments were performed inside a Faraday cage and at room temperature (23 ± 1 °C).
:3 H2O2:H2SO4, reacts violently with many organic materials and should be used with extreme care; it should not be stored in a sealed container). A platinum wire and a silver wire (0.5 mm diameter) were used as the counter electrode and the quasi-reference electrode, respectively. They were mechanically polished prior to use to remove the oxide film, rinsed with acetone and then dried under vacuum. The distance from the tip of the two electrodes to the center of the working electrode was set at 2 mm to minimize the effects of uncompensated electrolyte resistance. All potentials were measured using Fc/Fc+ (50 μM ferrocene in ILs) as the internal reference. Prior to the measurements, the entire system was dried in a vacuum oven at 60 °C for at least 2 h to remove any water and other impurities. The total gas flow was fixed at 200 sccm by digital mass flow controllers (MKS Instruments). Two mass flow controllers were used to adjust the volume ratio of the test gas flows. The highly purified gases (N2 and O2) were used containing <10 ppm water as described by the manufacturers. All the gas analytes were prepared by pre-mixing the gases in a 500 mL glass bottle with a stirring fan and then connecting to a drying column consisting of silica gel and solid calcium chloride before their introduction into the electrochemical cell. Using this drying process, the water level could be limited to <10 ppm. The gas was run for at least 12 h to ensure that equilibrium was established before the measurements were made. The IL system was purged constantly during the experiments with dry nitrogen or a mixture of nitrogen and oxygen to prevent any trace amounts of water or other impurities. All experiments were performed inside a Faraday cage and at room temperature (23 ± 1 °C).
EQCM measurements
The electrical admittance curve of the QCM was measured with an Agilent 4395A network/spectrum/impedance analyzer (Agilent Technologies, USA). Simultaneous electrochemical measurements were made using a CHI 1000A electrochemical workstation (CH Instruments, USA). A 1 μF capacitor was connected between the working electrode and the Agilent impedance analyzer to minimize the instrumental interference between the potentiostat and the Agilent impedance analyzer. The QCM was allowed to resonate for at least 2 h at the initial conditions for a baseline signal and then various potential programs were applied for the electrochemical measurements. The Agilent analyzer was preferred over other routinely used frequency measuring devices (e.g. RQCM) for QCM data monitoring because the IL–electrode interface is much more viscous than traditional aqueous electrolytes. In such a scenario, damping of the oscillation wave is so high that a more sophisticated instrument is required to maintain the crystal oscillation and obtain the correct data.Results and discussion
Generation of superoxide anion radicals in ILs
The superoxide radical was first generated by the electrochemical reduction of oxygen in all three ILs ([BMIM][NTf2], [BdMIM][NTf2] and [BMPY][NTf2]) and was examined at the Au QCM electrode by CV to analyze the stability of the ILs. Fig. 1 shows that the resulting peaks for oxygen reduction and superoxide oxidation were observed at negative potentials compared with those in a nitrogen atmosphere (inset). The cathodic peak between −1.2 and −1.3 V was assigned to the one-electron reduction of O2 to O2˙− whereas the anodic peak between −1.2 and −1.0 V was the subsequent oxidation of O2˙−:| O2 + e− ⇌ O2˙− | (1) |
 | ||
| Fig. 1 Second potential cycle cyclic voltammograms of the three ILs in the absence and presence of 40 vol% oxygen. Inset: enlarged scale in the absence of oxygen. Scan rate = 100 mV s−1. Nitrogen was used as the dilution gas. | ||
The presence of the O2˙− oxidation peak provides evidence that the O2˙− generated in situ is chemically stable within the time-scale of experiments in the three ILs studied. These results are consistent with those reported previously.22,26,37–40
It is necessary to determine the concentration of O2 in each IL for quantitative evaluation. In this study, the concentration of O2 (C) and its diffusion coefficient (D) were determined by a chronoamperometric technique with both macro- and ultra-micro disk electrodes. For the macro-disk electrode, when a sufficient over-potential is applied, the current density is inversely proportional to the square root of time, as represented by Cottrell's equation:
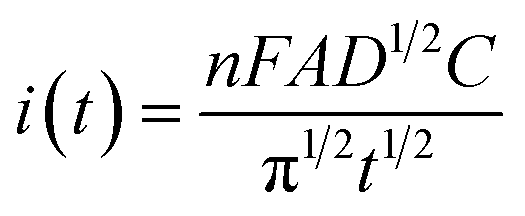 | (2) |
| iss = 4nFDCr0 | (3) |
It is clear from Table 1 that the order of magnitude for the oxygen concentration is [BdMIM][NTf2] > [BMPY][NTf2] > [BMIM][NTf2]. However, as can be seen from the results of the EQCM experiments, [BdMIM][NTf2], with the highest oxygen concentration, has the lowest peak current for superoxide oxidation. The frequency change in this IL was also the smallest among the three ILs studied. This indicates that, in this viscous electrolyte system, D might play a more significant part in the ORR than the concentration. For this reason, we addressed the contributions from D in more detail. As shown in Fig. 1, the cathodic reduction peak current (ipc) in [BdMIM][NTf2] is lower than that in the other two ILs, as it has an extremely small D value, close to one-quarter of that in [BMIM][NTf2]. The similarity of ipc in both [BMPY][NTf2] and [BMIM][NTf2] is a result of their close values of D and C. The solubility of oxygen in [BMPY][NTf2] is higher than that in [BMIM][NTf2], but the diffusion of O2 in [BMIM][NTf2] is faster. Note that the order of the anodic superoxide oxidation peak current (ipa) in the three ILs is different from ipc, with the following order: ipa,[BMPY][NTf2] > ipa,[BMIM][NTf2] > ipa,[BdMIM][NTf2]. The anodic peak potential (Epa) also shows a significant difference. For example, Epa in [BMIM][NTf2] is shifted by about 300 mV positive compared with other two ILs. The change in the superoxide oxidation processes can be explained as an ECrev mechanism in which one-electron oxygen reduction is followed by the reversible formation of an ion pair O2˙−⋯[Cation]+ as proposed by Compton and coworkers:26,27
| O2 + e− ⇌ O2˙− + [Cation]+ ⇌ O2˙−⋯[Cation]+ | (4) |
The rapid ion pairing of O2˙−⋯[Cation]+ hampers the superoxide oxidation process, causing a positive shift in the potential and leading to an increased peak-to-peak separation (ΔEp), making it more electrochemically irreversible than expected for a simple E process. The extent of irreversibility depends on the kinetics of the coupled chemical processes.
Experiments with the O2/O2˙− couple were performed in three ILs at varying scan rates (detailed data are given in Fig. S1†) to further investigate the relationship between the stability of the superoxide and the structure of the ILs. Fig. 2 shows the variations in ipc, ipa, Epc, Epa and ΔEp when the scan rates were varied. The peak currents were proportional to the square root of the scan rate for all three ILs. This shows that the reduction of O2 and the oxidation of O2˙− in the ILs are diffusion-controlled. Epc shifted to a more negative value and Epa shifted to a more positive value with increasing scan rates. This is characteristic of a quasi-reversible system. The Epa shows notable differences among the three different ILs studied, but Epc remains at a relatively constant value, reflecting the different degrees of association between the electro-generated superoxide and the cation. The uncompensated resistance (iRu) and surface reaction kinetics at the metal electrodes were used to explain the variation in the ΔEp of the O2/O2˙− redox couple in different aprotic solvents.37,43 However, the ΔEp values of the oxygen redox processes in these three ILs follows the order: ΔEp,[BMIM][NTf2] ≫ ΔEp,[BdMIM][NTf2] > ΔEp,[BMPY][NTf2]. The broadest ΔEp peak can be observed in [BMIM][NTf2], but the uncompensated resistance (iRu) in [BMIM][NTf2] should be the lowest as its conductivity is the highest of the three ILs. Thus the variation in ΔEp is mainly attributed to the variation in the electrode reaction kinetics in different ILs and there may be an effect from iRu, although this is too small to have a significant effect. The variation in electrode reaction kinetics were the results of the (bond-forming) ion paring O2˙−⋯[Cation]+, which leads to a positive shift in Epa as well as an increase in ΔEp. The stronger the interactions between O2˙− and the cation, the larger the value of ΔEp. The formation of the ion pair is probably a result of both electrostatic interactions and hydrogen bonding between the O2˙− and the [Cation]+. It has long been known that all three of the imidazolium ring protons (C2, C4 and C5) are acidic, although the proton in the C2 position is the most acidic. [BdMIM]+ has fewer protons at the C2 position of the ring, which makes it a weaker hydrogen bond donor than [BMIM]+. As the ability to donate hydrogen bonds increases with the increasing s-character of the C–H bond, imidazolium-based ILs commonly have a higher hydrogen bond acidity than pyrrolidinium- and alkyl ammonium-based ILs.
 | ||
| Fig. 2 Plot of (A) ipc and ipa and (B) Epc, Epa and ΔEp versus the scan rate in three ILs in the presence of oxygen (40 vol%) at the second potential cycle. The dilution gas was nitrogen. | ||
Multiple cycles of CV to characterize superoxide stability in ILs
The kinetics of forming O2˙−⋯[Cation]+ ion pairs has been reported as a reversible reaction in which O2˙− can be stabilized in ILs. Several groups have identified that such ion pairing promotes the O2˙− species to eventually abstract protons from the imidazolium cation to form hydrogen peroxide and a new product:32,33
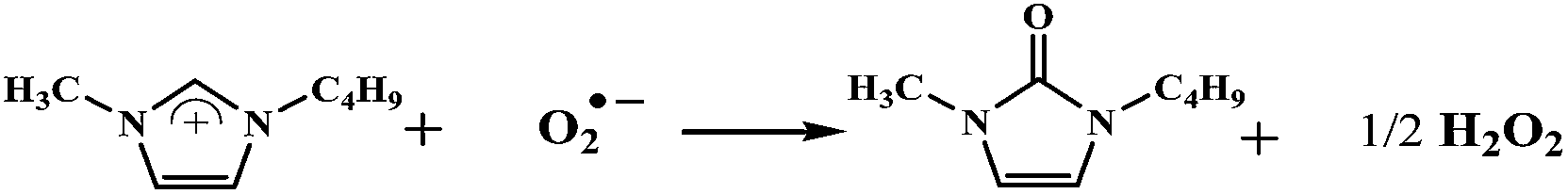 | (5) |
 | (6) |
Note that these reactions are irreversible, thus the long-term stability of ILs against this radical anion still needs to be investigated. For this purpose, the reversibility of the O2/O2˙− redox couple in the ILs was further investigated by multiple cycles of CV at various scan rates.
Fig. 3 shows the data for the continuous cycling (20 cycles) of the oxygen redox process in three ILs at fast (500 mV s−1) and slow (10 mV s−1) scan rates (other scan rates are shown in Fig. S2†). The ipc gradually decreased with increasing numbers of scan cycles. This is explained based on two factors: (1) the decrease in the double-layer charging current in subsequent cycles; and (2) the depletion of oxygen as a result of the redox processes of O2/O2˙−, as oxygen cannot be rapidly supplied from the bulk ILs to the electrode interface. Several interesting trends were observed for ipa. For example, ipa remains almost constant in both [BMIM][NTf2] and [BMPY][NTf2] at 500 mV s−1. It decreases with each subsequent scan cycle in [BdMIM][NTf2]. A small intermediate peak at about −0.7 for the oxidation of O2˙− and hydrogen peroxide (H2O2) to O2 is observed; a similar phenomenon has been reported by other workers.32 At a scan rate of 10 mV s−1, the ipa in [BMIM][NTf2] and [BdMIM][NTf2] dramatically decreases, whereas ipa decreases very slowly in [BMPY][NTf2]. In addition, compared with a scan rate of 500 mV s−1, a new small anodic peak at about −0.4 V at a scan rate of 10 mV s−1 was observed in [BMIM][NTf2] (Fig. 3D), which is assigned to the oxidation of HO2−.32 The enlarged view of this potential region in three ILs is shown in Fig. S3.† A smaller peak at about −0.4 V can be observed in [BdMIM][NTf2], but is hardly seen in [BMPY][NTf2], re-confirming that the abstraction of protons from [BMIM][NTf2] is easiest among the three selected ILs, followed by [BdMIM][NTf2] and [BMPY][NTf2].
 | ||
| Fig. 3 Twenty typical cycles of CV at different scan rates of (A–C) 500 mV s−1 and (D–F) 10 mV s−1 in three ILs in the presence of oxygen (40 vol%). Inset: CV in nitrogen without oxygen. Nitrogen was used as the dilution gas. | ||
Therefore the reversibility of the O2/O2˙− redox couple in the three ILs is different under different conditions (scan rate, cycle number), suggesting that the stability of the superoxide depends on the structure of the ILs. The significant irreversibility observed in the multiple CV experiments at slower scan rates indicates that the kinetics of the ion pairing and subsequent chemical reactions are slow, which can be observed more clearly at slower scan rates. At slow scan rates some of the O2˙−⋯[Cation]+ ion complex can form H2O2 and other compounds (e.g. 2-imidazolones). Thus a mechanism of homogeneous chemical reactions (including reversible ion pair formation and an irreversible protonation process) coupled to the electron transfer process of oxygen can be summarized as follows:
 | (7) |
The quantitative diagnosis of chemical reactions coupled with electrode reactions is often based on the relative current heights of the anodic and cathodic peaks. If the conversion of the ion pair complex to H2O2 is a slow process, at fast potential scan rates most of the O2˙− will be oxidized back to O2, thus the value of ipa/ipc will be close to 1. If the conversion of the ion pair complex to H2O2 is fast, some of the O2˙− will be lost to this process. Less O2˙− will be available for oxidation back to O2 on the backward potential scan and therefore the anodic peak will be smaller and the value of ipa/ipc will be <1. The ipa/ipc values of three ILs at the fifth cycle were plotted as a function of scan rate (Fig. 4). Note that the ipa and ipc values were achieved using the CHI 1040A electrochemical analyzer software after subtraction of the background current. In addition, the main anodic peak of about −1.1 V is about 13 times larger than that of the peak at about −0.7 V in [BdMIM][NTf2]. Therefore we believe that the effect of this intermediate peak is very small to be negligible. The trends of the plots are similar for [BdMIM][NTf2] and [BMIM][NTf2] in which ipa/ipc decreases at fast scan rates with increasing scan rate, although, at slow scan rates, ipa/ipc increases with increasing scan rates. The value of ipa/ipc showed the least change in [BMPY][NTf2], but a slight decrease in ipa/ipc can still be observed at scan rates <20 mV s−1, even in this IL.
 | ||
| Fig. 4 Variations in the ipa/ipc for the fifth cycle with different scan rates in three ILs in the presence of oxygen (40 vol%). Nitrogen was used as the diluting gas. | ||
Nicholson and Shain44 reported that the general trend followed by the peak current ratio as a function of scan rate can serve as a quick diagnosis for electrochemical mechanisms involving coupled chemical reactions. At fast scan rates the oxygen reduction process is an ErCr mechanism with a reversible electron transfer (Er) followed by a reversible chemical reaction (Cr) (eqn (4)). At slow scan rates the reaction is changed to a mechanism of a reversible electron transfer (Er) followed by an irreversible chemical (Ci) reaction (ErCi mechanism), corresponding to ion pair formation and further protonation processes during the following superoxide reaction in the IL (eqn (7)). Fig. 5 compares the change in ipa/ipc at various scan rates with the increase in the number of cycles in three ILs. The change in the ipa/ipc value in ILs at different scan cycles is an indicator of the loss in the reversibility of the redox process with time. The smallest change in ipa/ipc observed in [BMPY][NTf2] indicates that the stability of the O2˙− in this IL is more than 100 min. The most striking result is in [BMIM][NTf2], where ipa/ipc shows a large change at 30 min at scan rates of 20 and 10 mV s−1 (if we convert the CV scans to a time-scale, the 20 cycles for these two scan rates are equivalent to 50 and 100 min, respectively). The change in ipa/ipc could be seen in [BdMIM][NTf2], although it is much smaller than that of [BMIM][NTf2], showing the varying stability of the superoxide radical in different ILs. In summary, the rate of reaction between the O2˙− and the cation of the ILs follows the order [BMIM][NTf2] > [BdMIM][NTf2] > [BMPY][NTf2]. This order is consistent with the result obtained from the ΔEp value.
 | ||
| Fig. 5 Variations in the values of ipa/ipc in each CV cycle at different scan rates in three ILs in the presence of oxygen (40 vol%). Nitrogen was used as the dilution gas. | ||
EQCM evaluation of the ORR in ILs
To further investigate the effect of oxygen reduction in ILs, an EQCM method was used. The QCM integrated with electrochemical methods enabled us to obtain dynamic QCM data as a function of time and as a function of the electrochemical processes, thereby providing a framework for understanding how the ionic structures and populations changed while the potential was scanned from the anodic to the cathodic directions and vice versa. The EQCM technique can thus trace the time-resolved variations at the IL–electrode interface that are often too subtle to be detected with spectroscopic techniques (i.e. measuring the orientation of the ions at specified potentials only). Using this analysis for our system, we could experimentally monitor changes in several physical parameters related to the IL–electrode interface during the electron transfer process of the O2/O2˙− couple and its subsequent chemical reactions. Fig. 6 shows the simultaneous responses of changes in potential, current, oscillation frequency (Δf0) and damping resistance (ΔR1) for [BMPY][NTf2], both in the presence and absence of oxygen as obtained from the EQCM experiments. A decrease in frequency during the oxygen reduction process in the cathodic potential scan and an increase in frequency during the superoxide oxidation process at the anodic potential scan were observed. However, no obvious change in frequency was observed under pure N2 conditions for same experimental conditions, suggesting that the potential polarization does not generate significant changes in the electrode–electrolyte interface. This kind of EQCM response is also observed during the electrode reaction of [Fe(CN)6]4− and [Fe(CN)6]3− in aqueous solution, which has been explained as small variations (<3%) in the density and viscosity of the depletion layer that accompany the oxidation or reduction of the electroactive species.45 We also observed this in [BMPY][NTf2] during the redox process of ferrocene (Fig. S4†). If we assume that the mass transport of O2 during its redox process is slow in the ILs, so that it contributes little to the frequency change, the frequency waves that are very closely related to the CV current curves should be assigned to the change in the viscosity and density of the electrolyte near the electrode surface resulting from molecular interactions between the superoxide and the ILs. At an applied negative potential, it has been reported that, in an IL, the electrode has a surface charge that is over-screened by a monolayer of counter ions (cations); a balancing excess of opposite ions (anions) exists in the next layer to counter the over-screening.46 The increase in applied potential also causes this effect to be spread out over a larger number of ion pairing layers.47,48 However, these rearrangements as a result of potential cycling between 0 and −1.6 V are fully reversible and do not cause obvious changes in viscosity near the electrode, reflected in the flat frequency curve obtained with nitrogen alone. There is a shift in frequency after the electron transfer process in the presence of oxygen, observable in all three ILs, but with different amplitudes of frequency changes (Fig. S5†). Thus the reduction of O2 to O2˙− in the IL results in a spectacular dissimilarity in the diffusion coefficients between O2 and O2˙−, undergoing large conformational or solvation changes after electron transfer. The difference in their interaction with the ILs contributes to the change in the density and viscosity of the depletion layer. | ||
| Fig. 6 Simultaneous recording of potential, current, Δf0 and ΔR1 during three cycles of CV in [BMPY][NTf2] in the absence and presence of oxygen (40 vol%). Scan rate = 10 mV s−1. Nitrogen was used as the dilution gas. The momentary and reversible decrease in frequency during the reduction reaction of oxygen is caused by the small variations at the electrode surface that accompany the redox process (the electroactive species may diffuse to the depletion layer from a balanced state to either an oxidized or reduced state). | ||
Martin et al.49 reported a series of equivalent circuit parameters and a modified Butterworth–Van Dyke equivalent electrical circuit for the characterization of a QCM with simultaneous mass and liquid loading. The values of |Δf0/ΔR1| allow the separation of the mass and viscosity effects for the frequency signals. For a 10 MHz quartz crystal, if |Δf0/ΔR1| is larger than 11.6 Hz Ω−1, then the frequency changes can be ascribed predominantly to the mass effect. In the case of oxygen reduction, for both the waves for a single cycle and the overall trend after three cycles, the |Δf0/ΔR1| ratios are smaller than 11.6 Hz Ω−1, confirming that the QCM response is mainly due to viscosity changes at the interface.
The O2˙− generated couples with the cation of an IL near the electrode surface to form an ion pair complex, which is not completely reversible, as discussed earlier, and is hypothesized to lower the local viscoelasticity near the electrode, leading to the change in oscillating frequency (Scheme 2). In general, by consideration of the similar structures in ILs, the differences in viscosity are mainly influenced by hydrogen bonding and van der Waals interactions.50,51 In this case, O2˙− as a hydrogen bonding acceptor interacts with the cation and competes with the anion for the proton. This leads to [Cation]⋯O2˙− ion pairing, thereby suppressing the hydrogen bonding in ILs. This is expected to reduce the viscosity as a result of weakened interactions between the cation and anion.50
 | ||
| Scheme 2 Schematic illustration of the ionic arrangement with (A) increasingly negative potential and (B) the formation of ion pairs in ILs. | ||
The decay length, δ, which describes the approximate thickness of the electrolyte and electrode interface, the effective change of which is sensed by the EQCM, is limited by a function of the density and viscosity of the solution as follows:45
| δ = η1/2/(πf0ρ)1/2 | (8) |
The EQCM response appears to be governed essentially by the cation in the diffusion layer near the negatively electrified electrode surface and the ion pairing force between the cation and O2˙−. Fig. 7A demonstrates simultaneously the recorded frequency changes over a total of 20 CV cycles, sweeping in the selected ILs in the presence of oxygen (40 vol%) at a scan rate of 20 mV s−1. In the range of selected ILs, we observed distinct variations in the changes in f0 generated by potential cycling. It is clearly seen that the order of magnitude for the total change in Δf0 is [BMIM][NTf2] (602.4 Hz) > [BMPY][NTf2] (303.4 Hz) > [BdMIM][NTf2] (178.4 Hz). A flat frequency curve was observed in [BMIM][NTf2] in the absence of oxygen, as in the other two ILs (data not shown), indicating that the QCM response can be ascribed to the ORR. [BMIM][NTf2] presents the highest change in f0 as it combines the maximum diffusion coefficient with the strongest intensity of ion pairing. [BdMIM][NTf2] presents the lowest f0 response because of its minimum diffusion coefficient dominates over the moderate ion pairing force. The numerical values of the resulting Δf0 before and after CV sweeping in each IL with different potential cycling scan rates are shown in Fig. 7B. Surprisingly, when the scan rate was higher than or equal to 100 mV s−1, suggesting that the oxygen reduction as well as the reactivity of the superoxide with ILs was reversible, the frequency was essentially unchanged. However, at slower scan rates, when there is a longer period of negative potential polarization causing more and more cation layers to be gathered near the electrode surface, an increase in the local concentration of the cation is expected, which can lead to a higher possibility of forming ion pairs and thus a higher EQCM response. This was exactly the case when the scan rates were less than or equal to 50 mV s−1, with significantly higher values of Δf0 as a result of the stronger and irreversible reactivity of the superoxide with the ILs. These results further confirmed our previous hypothesis that the QCM response during oxygen reduction comes from the viscosity and density changes resulting from the formation of the ion pair complex. The smaller dependence between the QCM response and the scan rate (>50 mV s−1) denies another possibility that QCM response comes from the mass loading, such as the adsorption of superoxide or other intermediate compounds, as more adsorbate will be produced at slower scan rates and therefore the frequency due to mass loading should decrease rather than increase. The other type of variations seen in the frequency curves is the kind of frequency wave produced during each cycle of the measurement, which provides an indication of the stability of the superoxide anion as related to the electrochemical data. This wave is clearly visible in the case of the ferrocene couple (Fig. S4†), where there is no overall increase in frequency and the direction of the frequency shift changes fairly coherently with the direction of the potential scan. This frequency wave is also observable for [BMPY][NTf2], even for the 20th cycle (Fig. 7A). This indicates that the superoxide is consistently being re-oxidized, lessening the ion pairing effect during the anodic scan and reversing the direction of the shift in frequency. In contrast, this kind of wave is only visible in the first few cycles for [BMIM][NTf2], after which only an increase in the overall frequency is seen. This shows that the O2˙− generated has more irreversible interactions within this IL. [BdMIM][NTf2] lies between these two, further supporting the results obtained via electrochemical measurements for the long-term stability of O2˙− in different ILs. Thus, by combining the two results, the long-term stability of the O2˙− in ILs can be determined in situ in several electrochemical reactions.
 | ||
| Fig. 7 (A) Simultaneously recorded frequency of 20-cycle CV scans in three ILs in the absence and presence of oxygen (40 vol%) at a scan rate of 20 mV s−1 and (B) total increase in frequency after 20 scan cycles at different scan rates (10–500 mV s−1) in three ILs in the presence of oxygen (40 vol%). Nitrogen was used as the dilution gas. The overall effect of the [Cation]⋯O2˙− ion pairing is a decrease in viscosity, thereby increasing the overall frequency of the QCM. This effect is irreversible and probably due to the interface reactions that led to the viscoelastic property changes at the Au–IL interface. | ||
Long-term stability of the IL–electrode interface in the presence of O2˙−
The long-term stability of ILs in the presence of electrochemically generated O2˙− was determined in independent experiments at a much longer time-scale than the CV experiments using the EQCM method. We performed the CV experiment for the ORR every day without changing the ILs and the EQCM data were collected. Fig. 8A shows the EQCM results of oxygen reduction in the ILs at the first day and at the 21st day. The ipa/ipc value in [BMIM][NTf2] significantly decreased to about 37.3 ± 1.6% from its initial value. This change is much bigger than that in [BMPY][NTf2] (15.1 ± 2.3%). Furthermore, the simultaneously recorded frequency shows significant variations (Fig. 8B). There is a noticeable difference in [BMIM][NTf2], reflecting a decrease of about 160 Hz in the frequency after potential cycling. However, the frequency curves almost completely overlapped in [BMPY][NTf2]. As discussed earlier, ion pairing may ultimately lead to the O2˙− species abstracting the proton from the IL cations, especially from the imidazolium-based ILs, to form carbine and H2O2. However, it has been shown that peroxide species can be oxidized to H2O in ILs. In this instance, H2O moisture as a proton source plays a greater part in the acceleration of the protonation reaction of the O2˙− species. The existence of a proton source leads to the spontaneously rapid protonation of the electro-generated superoxide species, thus lowering the ion pairing effect and the reversibility of the O2 redox reaction. These results demonstrate that ILs containing [BMPY] offer a superior long-term stability for O2˙− attack than imidazolium salts. | ||
| Fig. 8 (A) Four-cycles of CV and (B) simultaneously recorded changes in Δf0 for [BMIM][NTf2] and [BMPY][NTf2] on the first day and after 3 weeks in the presence of oxygen (40% v/v). Scan rate = 20 mV s−1. Nitrogen was used as the dilution gas. | ||
Conclusions
The ORR frequently interferes with electrochemical energy conversion reactions and sensors and is also a critical factor in the use of ILs for such applications. This is a result of its potential to disrupt the stability of ILs and to modify the reaction mechanisms. We investigated the ORR in this new class of electrolytes by an EQCM approach in three structurally different ILs. Our results showed that the electro-generated O2˙− couples with the cations of the ILs to form ion pair complexes. The reversibility of the O2/O2˙− reaction, which reflects the extent of the formation of ion pairs, decreases at slow scan rates. The degree of ion pairing strongly depends on the cation of the ILs. The [BMIM]-based ILs tend to form the strongest ion pairs with the superoxide radical, followed by the [BdMIM]- and [BMPY]-based ILs. The formation of the [Cation]⋯O2˙− ion pairs was proved to lower the local viscosity of the electrolyte near the electrode, reflected by the oscillating frequency change of the QCM measurements. The stability of the ILs in the presence of the in situ generation of O2˙− was characterized. It was found that the IL containing [BMPY] offered a superior long-term stability for O2˙− attack compared with those based on imidazolium salts.The principles obtained from this work can be extended to other redox processes in ILs. The results from this study allow the relationship between the structure of ILs and their chemical stability towards O2˙− to be characterized, which is important for the selection and design of new and stable electrolytes and for further utilization of O2˙− in many applications. This study also established that EQCM is an effective tool to investigate the redox reaction at the IL–electrode interface. Work is in progress to use the EQCM technique to study different types of electron transfer reaction in various ILs.
Acknowledgements
X. Zeng thanks the Office of Naval Research for support (Grant N000141010734).References
- A. Rehman and X. Q. Zeng, Acc. Chem. Res., 2012, 45, 1667–1677 CrossRef CAS PubMed.
- J. N. Barisci, G. G. Wallace, D. R. MacFarlane and R. H. Baughman, Electrochem. Commun., 2004, 6, 22–27 CrossRef CAS PubMed.
- J. Ding, D. Zhou, G. Spinks, G. Wallace, S. Forsyth, M. Forsyth and D. MacFarlane, Chem. Mater., 2003, 15, 2392–2398 CrossRef CAS.
- S. T. Handy, Chem.–Eur. J., 2003, 9, 2938–2944 CrossRef CAS.
- M. P. Jensen, J. Neuefeind, J. V. Beitz, S. Skanthakumar and L. Soderholm, J. Am. Chem. Soc., 2003, 125, 15466–15473 CrossRef CAS PubMed.
- P. Wang, S. M. Zakeeruddin, J.-E. Moser and M. Grätzel, J. Phys. Chem. B, 2003, 107, 13280–13285 CrossRef CAS.
- C. Yang, Q. Sun, J. Qiao and Y. Li, J. Phys. Chem. B, 2003, 107, 12981–12988 CrossRef CAS.
- S. M. Zakeeruddin and M. Grätzel, Adv. Funct. Mater., 2009, 19, 2187–2202 CrossRef CAS.
- C. J. Allen, S. Mukerjee, E. J. Plichta, M. A. Hendrickson and K. M. Abraham, J. Phys. Chem. Lett., 2011, 2, 2420–2424 CrossRef CAS.
- V. Chakrapani, F. Rusli, M. A. Filler and P. A. Kohl, J. Phys. Chem. C, 2011, 115, 22048–22053 CAS.
- S.-L. Chou, J.-Z. Wang, J.-Z. Sun, D. Wexler, M. Forsyth, H.-K. Liu, D. R. MacFarlane and S.-X. Dou, Chem. Mater., 2008, 20, 7044–7051 CrossRef CAS.
- Y. Lauw, M. D. Horne, T. Rodopoulos, A. Nelson and F. A. M. Leermakers, J. Phys. Chem. B, 2010, 114, 11149–11154 CrossRef CAS PubMed.
- M. Anouti, L. Timperman, M. el hilali, A. Boisset and H. Galiano, J. Phys. Chem. C, 2012, 116, 9412–9418 CAS.
- T. Y. Kim, H. W. Lee, M. Stoller, D. R. Dreyer, C. W. Bielawski, R. S. Ruoff and K. S. Suh, ACS Nano, 2010, 5, 436–442 CrossRef PubMed.
- M. C. Buzzeo, R. G. Evans and R. G. Compton, ChemPhysChem, 2004, 5, 1106–1120 CrossRef CAS PubMed.
- D. R. MacFarlane, M. Forsyth, P. C. Howlett, J. M. Pringle, J. Sun, G. Annat, W. Neil and E. I. Izgorodina, Acc. Chem. Res., 2007, 40, 1165–1173 CrossRef CAS PubMed.
- D. R. MacFarlane, J. M. Pringle, P. C. Howlett and M. Forsyth, Phys. Chem. Chem. Phys., 2010, 12, 1659–1669 RSC.
- M. J. A. Shiddiky and A. A. J. Torriero, Biosens. Bioelectron., 2011, 26, 1775–1787 CrossRef CAS PubMed.
- C. H. Xiao and X. Q. Zeng, J. Electrochem. Soc., 2013, 160, H749–H756 CrossRef CAS PubMed.
- M. C. Buzzeo, O. V. Klymenko, J. D. Wadhawan, C. Hardacre, K. R. Seddon and R. G. Compton, J. Phys. Chem. A, 2003, 107, 8872–8878 CrossRef CAS.
- M. Hayyan, F. S. Mjalli, I. M. AlNashef and M. A. Hashim, Int. J. Electrochem. Sci., 2012, 7, 9658–9667 CAS.
- M. Hayyan, F. S. Mjalli, M. A. Hashim and I. M. AlNashef, Ind. Eng. Chem. Res., 2012, 51, 10546–10556 CrossRef CAS.
- H. Gampp and S. J. Lippard, Inorg. Chem., 1983, 22, 357–358 CrossRef CAS.
- D. T. Sawyer, G. Chiericato, C. T. Angelis, E. J. Nanni and T. Tsuchiya, Anal. Chem., 1982, 54, 1720–1724 CrossRef CAS.
- D. T. Sawyer and J. S. Valentine, Acc. Chem. Res., 1981, 14, 393–400 CrossRef CAS.
- A. S. Barnes, E. I. Rogers, I. Streeter, L. Aldous, C. Hardacre, G. G. Wildgoose and R. G. Compton, J. Phys. Chem. C, 2008, 112, 13709–13715 CAS.
- E. I. Rogers, X.-J. Huang, E. J. F. Dickinson, C. Hardacre and R. G. Compton, J. Phys. Chem. C, 2009, 113, 17811–17823 CAS.
- M. M. Islam and T. Ohsaka, J. Phys. Chem. C, 2008, 112, 1269–1275 CAS.
- D. Méry, J. R. Aranzaes and D. Astruc, J. Am. Chem. Soc., 2006, 128, 5602–5603 CrossRef PubMed.
- T. L. Amyes, S. T. Diver, J. P. Richard, F. M. Rivas and K. Toth, J. Am. Chem. Soc., 2004, 126, 4366–4374 CrossRef CAS PubMed.
- A. Marcinek, J. Zielonka, J. Gȩbicki, C. M. Gordon and I. R. Dunkin, J. Phys. Chem. A, 2001, 105, 9305–9309 CrossRef CAS.
- M. M. Islam, T. Imase, T. Okajima, M. Takahashi, Y. Niikura, N. Kawashima, Y. Nakamura and T. Ohsaka, J. Phys. Chem. A, 2009, 113, 912–916 CrossRef CAS PubMed.
- I. M. AlNashef, M. A. Hashim, F. S. Mjalli, M. Q. A.-h. Ali and M. Hayyan, Tetrahedron Lett., 2010, 51, 1976–1978 CrossRef CAS PubMed.
- A. R. Hillman, J. Solid State Electrochem., 2011, 15, 1647–1660 CrossRef CAS PubMed.
- L. Yu, Y. Huang, X. Jin, A. J. Mason and X. Zeng, Sens. Actuators, B, 2009, 140, 363–370 CrossRef CAS PubMed.
- H. Y. Lee, J. B. Issa, S. S. Isied, E. W. Castner, Y. Pan, C. L. Hussey, K. S. Lee and J. F. Wishart, J. Phys. Chem. C, 2012, 116, 5197–5208 CAS.
- S. O'Toole, S. Pentlavalli and A. P. Doherty, J. Phys. Chem. B, 2007, 111, 9281–9287 CrossRef PubMed.
- M. T. Carter, C. L. Hussey, S. K. D. Strubinger and R. A. Osteryoung, Inorg. Chem., 1991, 30, 1149–1151 CrossRef CAS.
- R. G. Evans, O. V. Klymenko, S. A. Saddoughi, C. Hardacre and R. G. Compton, J. Phys. Chem. B, 2004, 108, 7878–7886 CrossRef CAS.
- Y. Katayama, H. Onodera, M. Yamagata and T. Miura, J. Electrochem. Soc., 2004, 151, A59–A63 CrossRef CAS PubMed.
- X.-J. Huang, E. I. Rogers, C. Hardacre and R. G. Compton, J. Phys. Chem. B, 2009, 113, 8953–8959 CrossRef CAS PubMed.
- Ionic Liquids Today-01-11.pdf from http://www.iolitec.de/en/Download-document/665-IonicLiquidsToday%2001-11.pdf.html.
- Oxygen Chemistry, ed. D. T. Sawyer, Oxford University Press, New York/Oxford, 1991 Search PubMed.
- R. S. Nicholson and I. Shain, Anal. Chem., 1964, 36, 706–723 CrossRef CAS.
- W. W. Lee, H. S. White and M. D. Ward, Anal. Chem., 1993, 65, 3232–3237 CrossRef CAS.
- R. M. Lynden-Bell, A. I. Frolov and M. V. Fedorov, Phys. Chem. Chem. Phys., 2012, 14, 2693–2701 RSC.
- R. Hayes, N. Borisenko, M. K. Tam, P. C. Howlett, F. Endres and R. Atkin, J. Phys. Chem. C, 2011, 115, 6855–6863 CAS.
- M. Z. Bazant, B. D. Storey and A. A. Kornyshev, Phys. Rev. Lett., 2011, 106, 046102 CrossRef.
- S. J. Martin, V. E. Granstaff and G. C. Frye, Anal. Chem., 1991, 63, 2272–2281 CrossRef CAS.
- P. Bonhôte, A.-P. Dias, N. Papageorgiou, K. Kalyanasundaram and M. Grätzel, Inorg. Chem., 1996, 35, 1168–1178 CrossRef PubMed.
- P. Hapiot and C. Lagrost, Chem. Rev., 2008, 108, 2238–2264 CrossRef CAS PubMed.
Footnotes |
| † Electronic supplementary information (ESI) available: Fig. S1–S6 as noted in the text are provided. See DOI: 10.1039/c5ra00396b |
| ‡ Present address: School of Science, Xi'an Jiaotong University, Xi'an, China. |
| This journal is © The Royal Society of Chemistry 2015 |