
Iron carbide and nitride via a flexible route: synthesis, structure and magnetic properties†
Abstract
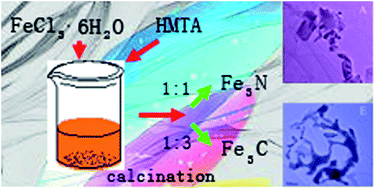
Single-phase Fe4N and Fe3C have been controllably synthesized from a coordination precursor by adjusting the molar ratio of hexamethylenetetramine to ferric chloride hexahydrate, both presenting good paramagnetic behavior, with maximum magnetizations that can reach 150.51 and 53.03 emu g−1 up to 17.5 kOe, respectively. The magnetic properties of Fe3C can be adjusted by the molar ratio of hexamethylenetetramine. Moreover, the structure, components and the possible mechanism were investigated carefully.
 Please wait while we load your content...
Please wait while we load your content...

