
Facile synthesis of silver-decorated reduced graphene oxide as a hybrid filler material for electrically conductive polymer composites
Abstract
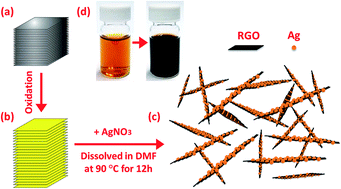
Nano silver-decorated reduced graphene oxide (Ag–RGO) sheets were synthesized by simply dissolving graphite oxide and silver nitrate in N,N-dimethylformamide and keeping the suspension at 90 °C for 12 h. These highly stable hybrid sheets were then incorporated into a polar polymer, polyvinylidene fluoride (PVDF), to prepare the Ag–RGO/PVDF nanocomposites via solution mixing. The Ag–RGO hybrid sheets were dispersed homogeneously in the polymer matrix, resulting in a low percolation threshold of 0.17 vol%. Above the percolation threshold, electrical conductivity of the Ag–RGO/PVDF composite system was about one order of magnitude higher than that of thermally reduced graphene/PVDF composites. This was attributed to the high intrinsic electrical conductivity of silver. The improved electrical properties render this novel composite system an attractive material for antistatic, electrostatic dissipative and electromagnetic/radio frequency interference shielding applications. Furthermore, the resistivity of the composite system increased with increasing temperature, generating a pronounced positive temperature coefficient effect of resistivity.
 Please wait while we load your content...
Please wait while we load your content...

