
Self-assembly of DNA wrapped carbon nanotubes and asymmetrical cyanine dyes into fluorescent nanohybrids†
Abstract
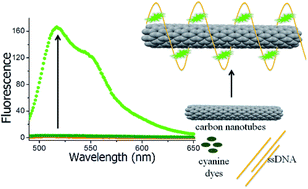
Light absorbing and emitting nanohybrid materials constructed from single stranded DNA, asymmetrical cyanine dyes and carbon nanotubes are presented. Asymmetrical cyanine dyes interact strongly with carbon nanotubes through noncovalent binding resulting in light absorbing nanoarrays for which the fluorescence emission is quenched. On the other hand, when carbon nanotubes are wrapped with single stranded DNA, a unique scaffold is formed on which asymmetrical cyanine dyes can self-assemble with increased quantum yields resulting in fluorescent nanohybrids. These three-component nanohybrid materials can absorb and emit light in desired ranges of the spectrum and are the first example where fluorescent dyes light up when bound to carbon nanotubes as opposed to being quenched. The DNA/carbon nanotube/dye nanohybrids are shown to be energetically favored, consistently forming self-assembled nanostructures that are obtained independent of the order of addition of components. These nanohybrid materials have been characterized for their binding stoichiometry and thermal stability by utilizing absorbance and fluorescence spectroscopy; furthermore, their fluorescence has been visualized with confocal microscopy. While the method presented here is valuable for fluorescent labeling of carbon nanotubes for biomedical applications, these nanohybrid materials are also potential candidates for photoactive bionanodevices as light harvesting systems and optical sensors.
 Please wait while we load your content...
Please wait while we load your content...

