
One-step electrochemical synthesis of Bi3.84W0.16O6.24 with superior photocatalytic activities†
Zuwei Song*,
Hui Dai and
Jiantao Tong
School of Chemistry and Pharmaceutical Science, Qingdao Agricultural University, Qingdao 266109, China. E-mail: cataszw@hotmail.com; Fax: +86 532 86080213; Tel: +86 532 86080374
First published on 13th February 2015
Abstract
Bi3.84W0.16O6.24 nanoparticles were successfully prepared by a facile electrochemical route at room temperature in just 10 minutes. The phase composition, morphology and crystallinity can be well controlled by manipulating the volume ratio of ethanol–water in the solvent and the electric current value. The as-prepared products with relatively uniform size of 25 nm show superior visible light photocatalytic activities, which are even higher than that of nest-like Bi2WO6 particles with a larger specific surface area synthesized by a hydrothermal method.
Bismuth tungstate compounds with good photoelectric properties are a kind of functional material widely applied in environmental purification and remediation.1,2 Among them, Bi2WO6 is well known and has been investigated extensively in the field of heterogeneous photocatalysis.3–5 Recently, due to its narrower band gap than Bi2WO6, the lesser studied Bi3.84W0.16O6.24 has gained more and more attention and been proven to be another promising visible light-driven photocatalyst.6,7 To date, the syntheses of Bi3.84W0.16O6.24 are mainly focused on the hydrothermal treatment. Liu et al. synthesized Yb3+ and Ho3+ co-doped Bi3.84W0.16O6.24 nanocrystals in a hydrothermal environment, and investigated its up-conversion luminescence property.8 Disk-like Bi3.84W0.16O6.24, displaying good photocatalytic degradation ability to bisphenol A (BPA) under simulated solar light irradiation, was prepared under hydrothermal condition by Wang et al.9 A microwave hydrothermal method10 was also adopted to prepare Bi3.84W0.16O6.24 nanocrystals in alkalescent precursor suspension, which showed distinctive octahedron morphology, but a lower photocatalyst efficiency than that of Bi2WO6 synthesized under the same condition. As we all know, the advantages of hydrothermal method is in its well control on the crystallinity and morphology of as-prepared products. However, the high temperature and long holding time along with serious waste of energy are essential. Thus, the exploration of a facile and energy saving approach to prepare Bi3.84W0.16O6.24 remains a challenging task so far.
Herein, for the first time, a novel electrochemical route based on anodic oxidation and dielectric breakdown was developed to synthesize Bi3.84W0.16O6.24 nanoparticles in aqueous solution at ambient temperature. Compared with the hydrothermal method, the electrochemical route present many advantages such as its simplicity, low energy cost, easy control and environmentally friendly. Moreover, the influences of ethanol quantity in the electrolyte and the electric current value on the phase composition, morphology and crystallinity of Bi3.84W0.16O6.24 were investigated carefully.
The experiment was performed in a double cell bath with cooling system, using two pieces of bismuth sheets (100 mm × 20 mm × 2 mm, 99.9% purity) as the electrodes. The typical electrolyte is prepared as follows: 10.6 g Na2WO4·2H2O and 3.2 g KOH were dissolved in 74 mL mixed solvent composed of distilled water and ethanol under magnetic stirring for 10 min, followed by the addition of 6 mL H2O2 (30 vol%). The volume ratio of ethanol–water was adjusted to 0, 0.1, 0.3, 0.5 and 0.7, respectively. The bismuth sheets were parallelly dipped into the electrolyte for 4 cm and connected with the outer DC supply. Then the experiment was carried out at an electric current value 3 A for 10 min. The precipitate generated in the anode region was harvested by centrifugation, washed by deionized water for several times and dried at 60 °C. The obtained samples were characterized by an X-ray diffractometer (D8 Advance, Germany), field emission scanning electron microscopy (JSM-7500, Japan), transmission electron microscopy (HT7700, Japan) and a spectrophotometer (TU1901, China), and Rhodamine B (RhB) was used to evaluate their photocatalytic activities (see ESI†). For comparison, the bulk Bi3.84W0.16O6.24 powders were prepared by calcining Bi2O3 and WO3 powders with a stoichiometric ratio11 at 850 °C for a holding time of 2 h.
Fig. 1 shows the XRD patterns of the samples prepared at 3 A with various volume ratio ethanol–water. It is obvious that the products' phase compositions are sensitive to the volume ratio R. Without adding ethanol into the electrolyte, the products are composed of the mixed phases of cubic Bi3.84W0.16O6.24 (JCPDS no. 43-0447) and orthorhombic Bi2WO6 (JCPDS no. 39-0256), where the peak intensities of the latter are higher than that of the former. The samples obtained at R = 0.1 and 0.3 also displays the mixed phase of Bi3.84W0.16O6.24 and Bi2WO6. With the increase of ethanol addition, the Bi3.84W0.16O6.24 phase's characteristic (111), (200) and (220) peaks increase, while the characteristic (131) and (020) peaks of Bi2WO6 phase notably decrease and eventually diminish at R = 0.5, which means the single Bi3.84W0.16O6.24 phase with high crystallinity is obtained (Fig. 1d). Those results indicate that the ethanol promotes the formation of Bi3.84W0.16O6.24 phase, and the phase composition can be tuned by controlling the quantity of ethanol in the electrolyte. Further increasing the ratio to 0.7, the diffraction peaks weaken dramatically but the single phase characteristic of cubic Bi3.84W0.16O6.24 is still kept, suggesting that excess ethanol would inhibit the crystallization and growth of Bi3.84W0.16O6.24 crystallites. Thus, electrochemical method is a facile and novel way to controllable synthesis of bismuth tungstate.
 | ||
| Fig. 1 XRD patterns of the samples obtained at various ratio of ethanol–water: (a) 0, (b) 0.1, (c) 0.3, (d) 0.5 and (e) 0.7. | ||
Fig. 2 shows the XRD patterns of the samples obtained with different electric current values at R = 0.7. Obviously, Bi3.84W0.16O6.24 can be easily obtained with the current values varied from 1 A to 4 A and their corresponding diffraction peaks become stronger and sharper with the elevation of current values. Those results indicate that a larger electric current value would be beneficial to promote the crystallinity of Bi3.84W0.16O6.24 crystallites. The second phase of Bi2WO6 began to appear at 5 A, where the largest sparks emerged on the interface between the anode surface and the solution. It is worth noting that at a much lower current 0.5 A, the obtained samples are indexed to be Bi (JCPDS no. 44-1246), Bi2O3 (JCPDS no. 27-0052) and Bi2O3 (JCPDS no. 27-0050), and there are no evident anodic sparks in the whole process, quite different from that of 1 A to 4 A where the large and continues sparks are achieved. Thus, it is suggested that the formation of bismuth tungstate can be initiated by the anodic oxidation of Bi according to eqn (1), followed by the breakdown of the generated Bi2O3 barrier film under an appropriate electric condition, companied with numerous micro-sparks appeared the working surface of the anode. Meanwhile, according to eqn (2) and (3), the reaction between Bi2O3 and WO42− at the interface anode/electrolyte began at high local temperature supplied by the anodic spark, while the whole solution still keeps at room temperature. For the cathode, the generation of H2 from the reduction of H2O is the main reaction. Different from the hydrothermal method, the active Bi source resulted from the oxidation of bismuth anode, and the driven force or the energy needed was afforded by the anodic sparks.
| 2Bi + 6OH− → Bi2O3 + 3H2O + 6e− | (1) |
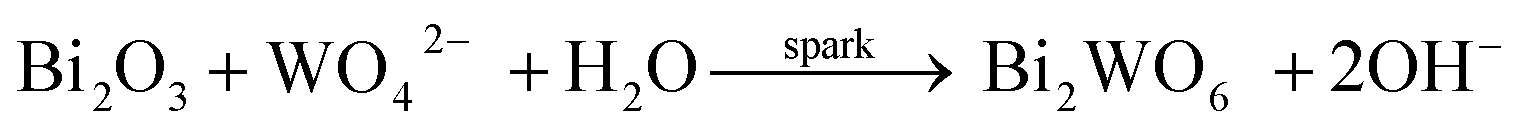 | (2) |
 | (3) |
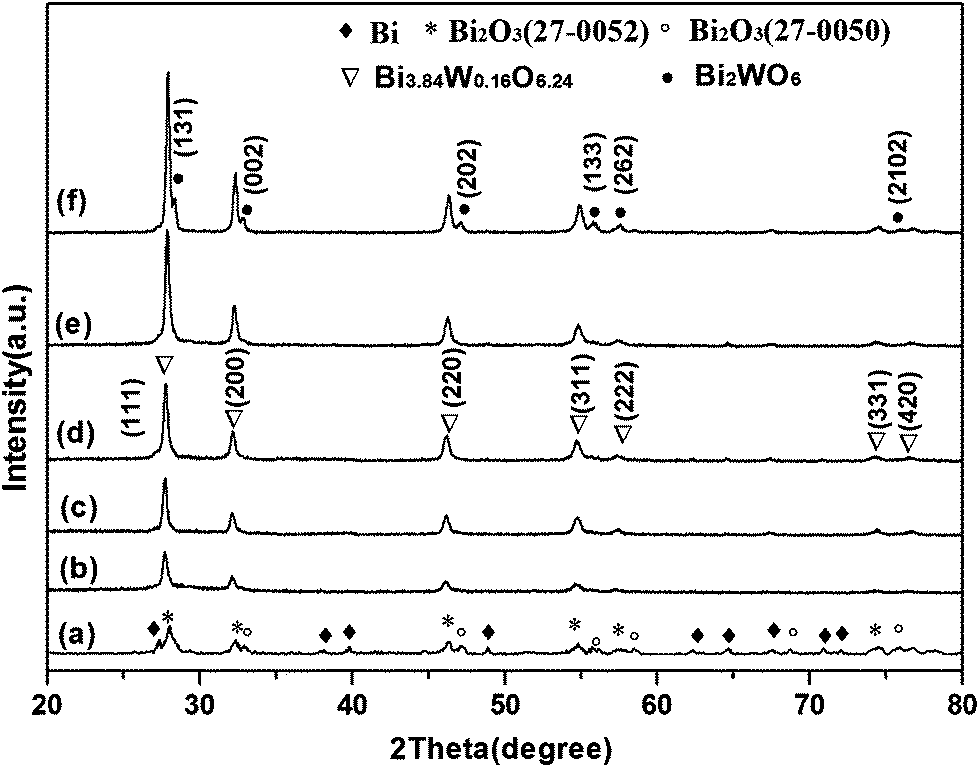 | ||
| Fig. 2 XRD patterns of the samples obtained at R = 0.7 with various electronic current values: (a) 0.5 A, (b) 1 A, (c) 2 A, (d) 3 A, (e) 4 A and (f) 5 A. | ||
Fig. 3 shows the representative SEM and TEM images of samples prepared at 3 A with different quantity of ethanol in the electrolyte. Bi3.84W0.16O6.24/Bi2WO6 composite prepared at R = 0.1 (BW-1) is consisted of two kinds of morphologies (Fig. 3a): one is regular nanoparticle with a little aggregation; the other is nanoflake with length of 200–300 nm, and most of which aggregated to larger and irregular flower-like shape. The nanoflakes are Bi2WO6 crystallites which tend to develop flake-like morphologies due to the intrinsic anisotropic crystallographic nature,12 and the flower-like aggregation derives from the fast nucleation and aggregation rates of Bi2WO6 nanoflakes. The single phase Bi3.84W0.16O6.24 obtained at R = 0.5 (BW-5) exists mainly in the form of nanoparticles with a wide size distribution of 20–100 nm (Fig. 3c). SEM observations confirm the inhibition of ethanol on the crystallization of Bi2WO6 phase, consistent with XRD results in Fig. 1. TEM image further confirmed the co-existence of nanoflakes and nanoparticles in the sample BW-1 (Fig. 3b), while the sole presence of nanoparticles with particle size of 20–100 nm in sample BW–5 (Fig. 3d). With the increase of R from 0.5 to 0.7, nanoparticles with relatively uniform size of 25 nm can be seen (BW-7, Fig. 3e). Moreover, the crystal lattice fringes of 0.325 nm in HR-TEM images (Fig. 3f) is in accordance with the (111) plane of Bi3.84W0.16O6.24.
 | ||
| Fig. 3 (a and b) are SEM and TEM images of sample BW-1; (c and d) are SEM and TEM images of BW-5; (e and f) are TEM and HRTEM images of BW-7. | ||
Based on the XRD results and SEM analyses, the amount of ethanol greatly influence the phase composition and morphology of bismuth tungstate. With the introduction of ethanol, the intensity and density of anodic sparks weakened gradually owing to the change of the conductivity and the dielectric constant of the electrolyte. Moreover, the activity of WO42− lowered as well due to the decrease of the solubility of WO42− in the mixed solvents13 and the formation of a WO42−–ethanol complex through hydrogen bonding between WO42− and ethanol.14 As the ratio R changed from 0 to 0.1 and 0.3, anodic sparks and WO42− ions available are enough for the generation and fast growth of the composite phases of Bi2WO6 and Bi3.84W0.16O6.24, while the appropriate synergy of sparks and WO42− ions make it possible for the generation and growth of single phase Bi3.84W0.16O6.24 with a wide size distribution (BW-5). In a milder solution with ratio R = 0.7, the further weakened anodic sparks and the lowered WO42− activity are still enough to the generation of Bi3.84W0.16O6.24 particles but unfavorable for their further development, and accordingly, the sample BW-7 with smaller crystallites is obtained.
To study the optical properties of the products, the UV-vis spectra of typical sample BW-1, BW-7 and the bulk Bi3.84W0.16O6.24 are collected and shown in Fig. 4. All the samples exhibit strong adsorptions in the visible region, exhibiting potential photocatalytic activity under visible light. The absorption edge of BW-7 is approximately at 490 nm, not only larger than that of the bulk one, but also larger than that of the previous report15 and the as-prepared Bi3.84W0.16O6.24/Bi2WO6 (460 nm). The results indicate that single Bi3.84W0.16O6.24 nanoparticles would exhibit an increased photo-absorption in the visible light region than that of composite one.
 | ||
| Fig. 4 UV-vis spectra of samples: (a) the bulk Bi3.84W0.16O6.24, (b) BW-1 and (c) BW-7. | ||
Fig. 5 shows the plots of RhB photodegradation as a function of irradiation time using sample BW-1, BW-7 and the bulk Bi3.84W0.16O6.24 powder. The blank test indicates that photo-induced self-sensitized photodegradation is negligible. The sample BW-7 shows superior photocatalytic activity, which is up to 96.5% degradation rate of RhB (Fig. 5a) in 180 min and much higher than that of BW-1 (55.0%, Fig. 5b) and bulk one (36.0%, Fig. 5c). The enhanced photocatalytic activity of BW-7 may be ascribed to the considerable higher specific surface area (41.6 m2 g−1, Fig. S1, ESI†) than that of BW-1 (10.7 m2 g−1, Fig. S1, ESI†) and the bulk Bi3.84W0.16O6.24 (1.6 m2 g−1, Fig. S1, ESI†). Moreover, it is noteworthy that the sample BW-7 also presents a higher activity than that of nest-like Bi2WO6 particles with a larger specific surface area (56.5 m2 g−1) synthesized by hydrothermal method in EDTA-mediated process,12 probably due to the narrower band gap of BW-7 (2.60 eV, Fig. 4) than that of Bi2WO6 (2.88 eV).
 | ||
| Fig. 5 Photodegradation of RhB under visible-light irradiation: (a) BW-7, (b) BW-1, (c) bulk Bi3.84W0.16O6.24 and (d) blank test without using catalysts. | ||
Conclusions
In summary, Bi3.84W0.16O6.24 nanoparticles with spherical shape and size of ca. 25 nm were rapidly synthesized by a simple electrochemical method in a mixed solvent with volume ratio of ethanol–water 0.7, exhibiting excellent photocatalytic activity under visible-light irradiation due to its high purity, large surface area and narrow energy gap. This work presents a new strategy to the synthesis of other Bi-based nanomaterials.Acknowledgements
This work was supported by the Natural Science Foundation of China (no. 21404066), the Natural Science Foundation of Shandong Province (no. ZR2012EMM017) and Scientific Research Foundation for Advanced Scholars of Qingdao Agricultural University (no. 631022).Notes and references
- L. Zhang and Y. Zhu, Catal. Sci. Technol., 2012, 2, 694–706 CAS.
- M. Qamar and A. Khan, RSC Adv., 2014, 4, 9542–9550 RSC.
- Z. Zhang and W. Wang, Dalton Trans., 2013, 12072–12074 RSC.
- Y. Fu, C. Chang, P. Chen, X. Chu and L. Zhu, J. Hazard. Mater., 2013, 254–255, 185–192 CrossRef CAS PubMed.
- Y. Zhang and Y. Xu, RSC Adv., 2014, 4, 2904–2910 RSC.
- X. Dai, Y. Luo, W. Zhang and S. Fu, Dalton Trans., 2010, 3426–3432 RSC.
- V. Nithya, R. Kalai Selvan, D. Kalpana, L. Vasylechko and C. Sanjeeviraja, Electrochim. Acta, 2013, 109, 720–731 CrossRef CAS PubMed.
- L. Liu, K. S. Yang, X. Y. Zhang, N. Qi, H. Li and Z. Zuo, J. Rare Earths, 2012, 30, 1092–1095 CrossRef CAS.
- C. Y. Wang, L. Y. Zhu, C. Song, G. Q. Shan and P. Chen, Appl. Catal., B, 2011, 105, 229–236 CrossRef CAS PubMed.
- S. S. Yao, J. Y. Wei, B. B. Huang, S. Y. Feng, X. Y. Zhang, X. Y. Qin, P. Wang, Z. Y. Wang, Q. Zhang, X. Y. Jing and J. Zhan, J. Solid State Chem., 2009, 182, 236–239 CrossRef CAS PubMed.
- A. P. Finlayson, E. Ward, V. N. Tsaneva and B. A. Glowacki, J. Power Sources, 2005, 145, 667–674 CrossRef CAS PubMed.
- L. Xu, X. Y. Yang, Z. Zhai and W. H. Hou, CrystEngComm, 2011, 13, 7267–7275 RSC.
- D. Wu, H. Zhu, C. Zhang and L. Chen, Chem. Commun., 2010, 46, 7250–7252 RSC.
- M. Shang, W. Z. Wang and H. L. Xu, Cryst. Growth Des., 2009, 9, 991–996 CAS.
- S. Chen, W. Tang, Y. Hua and X. Fu, CrystEngComm, 2013, 15, 7943–7950 RSC.
Footnote |
| † Electronic supplementary information (ESI) available. See DOI: 10.1039/c5ra00056d |
| This journal is © The Royal Society of Chemistry 2015 |