
Covalently functionalized graphene with D-glucose and its reinforcement to poly(vinyl alcohol) and poly(methyl methacrylate)
Lu Gan,
Songmin Shang*,
Chun Wah Marcus Yuen and
Shou-xiang Jiang
Institute of Textiles and Clothing, The Hong Kong Polytechnic University, Hong Kong. E-mail: shang.songmin@polyu.edu.hk; Fax: +852 2773 1432; Tel: +852 3400 3085
First published on 19th January 2015
Abstract
Chemically functionalized graphene has been synthesized by covalently grafting D-glucose on graphene for the first time through an esterification reaction. The D-glucose functionalized graphene was further applied in the preparation of poly(vinyl alcohol) and poly(methyl methacrylate) nanocomposites. The thermal and mechanical properties of the prepared nanocomposites were then investigated. It was demonstrated that the D-glucose was grafted on the surface of the graphene through a covalent attachment. The chemically functionalized graphene had better dispersibility in both water and dimethylformamide than the pristine graphene. The better dispersion of the functionalized graphene in both aqueous and organic solvents simplified the preparation of the polymer nanocomposites. It was found that the D-glucose grafted graphene dispersed homogeneously in both poly(vinyl alcohol) and poly(methyl methacrylate) matrices, increasing the thermal and mechanical properties of the polymers at the same time. The phenomena were ascribed to the introduction of the D-glucose, which induced strong hydrogen bonding interactions between the functionalized graphene and the polymers. The results of this study indicate that this is an effective approach for preparing well-dispersed graphene-based polymer nanocomposites by covalently attaching functional molecules on the surface of graphene.
1. Introduction
In recent years, nano-particle filled polymer composites have been intensively studied.1–4 Among these nano-particle fillers, the graphene related materials have received tremendous research interest for their distinct mechanical, thermal, optical and electrical properties.5,6 With a 2D single layered honeycomb structure, graphene is viewed as the thinnest, stiffest and strongest material in the world. Graphene-based composites also have found applications in various research areas such as supercapacitors,7 catalysts,8 and sensors.9 To achieve promising properties for the graphene-based polymer composites, graphene sheets are required to be dispersed homogeneously within the matrices. However, it is very difficult to obtain well exfoliated graphene sheets with single or few layers in both solvents and polymers due to the strong intermolecular van der Waals forces existing among them.10 Therefore, unless some pre-treatments to the graphene are made, it is almost impossible to prevent the aggregation of graphene sheets. Physical blending or adsorbing some molecules, such as surfactants11 and polymers,12 on the surface of the graphene sheets could effectively improve graphene dispersion. These foreign molecules act as the compatibilizer to weaken the van der Waals forces among the graphene sheets. Moreover, these compatibilizers more or less attenuate the reinforcing effect of the graphene, despite the fact that individual graphene sheets can be observed in the nanocomposites.For this reason, researchers are exploring the possibility of chemical functionalization of graphene. As a significant precursor and derivative of graphene, graphene oxide (GO) tends to be more compatible with some solvents13 and polymers14 because GO consists of many oxygen containing groups compared with graphene.15 These groups further facilitate the chemical modifications on the GO. Through simple organic reactions, the GO is able to covalently bind with some small molecules16 or polymers,17,18 and these chemically functionalized graphene (CFG) molecules are more dispersible in aqueous19 and organic20 solvents, and polymer matrixes.21 Moreover, the CFGs are more easily incorporated into the polymer without losing the inherent properties of the CFGs and the prepared nanocomposites.22 Recently, considerable interest has arisen towards synthesizing solvent dispersible or soluble CFGs.23,24 Long time stable CFG dispersions simplify the fabrication of the polymer composites through solvent mixing approaches.
As a most common and simple monosaccharide, D-glucose has been widely used in biology, pharmacy and clinically related areas for centuries.25 It is also the basic nutrient for almost all organisms. More significantly, D-glucose has numerous hydroxyl and aldehyde groups along its backbone and is soluble in water and many organic solvents such as dimethylformamide (DMF) and pyridine. If D-glucose could be covalently grafted on the graphene,26 the D-glucose grafted CFG is expected to have better dispersion in both aqueous and organic solvents, which is very important for preparing polymer nanocomposites. Compared with those CFGs, which can only be dispersed in either aqueous or organic solvents, the D-glucose grafted CFG has wider application potential for more polymer matrices. Moreover, this D-glucose grafted CFG is expected to have stronger interactions with the polymers, especially those with oxygen containing groups, such as poly(vinyl alcohol) (PVA) and poly(methyl methacrylate) (PMMA), because the existence of the D-glucose will facilitate the hydrogen bonding interactions between the CFG and the polymers.
Herein, we report for the first time, the attachment of graphene and D-glucose through covalent grafting. The D-glucose grafted graphene, named Glu-G, was further incorporated into PVA and PMMA to prepare their nanocomposites. Fourier transform infrared (FTIR) spectroscopy, thermogravimetric analysis (TGA), Raman spectroscopy and X-ray diffraction (XRD) were used to confirm the structure of the Glu-G. The dispersions of the Glu-G in water and DMF were also investigated. To study the reinforcing effect of Glu-G to polymers and the interaction between the fillers and the matrices, PVA/Glu-G and PMMA/Glu-G nanocomposites were characterized by morphological, mechanical and thermal properties.
2. Experimental
2.1 Materials
Natural graphite flake, hydrazine hydrate (N2H4·H2O), 4-dimethyl aminopyridine (DMAP, 99%), and N,N-dicyclohexylcarbo-diimide (DCC) were purchased from Aldrich (America). PVA (Mw ∼ 70![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) 000–85000, ∼87–89% alcoholized) was purchased from Accuchem (Canada). PMMA (mass density 1.19 g cm3, hardness ∼ 148 N mm−2) was purchased from International Laboratory (America). D-glucose was purchased from Unichem (China). All the other agents and solvents were of analytical grade and used as received. Deionized distilled water (DDW) was used exclusively in this study.
000–85000, ∼87–89% alcoholized) was purchased from Accuchem (Canada). PMMA (mass density 1.19 g cm3, hardness ∼ 148 N mm−2) was purchased from International Laboratory (America). D-glucose was purchased from Unichem (China). All the other agents and solvents were of analytical grade and used as received. Deionized distilled water (DDW) was used exclusively in this study.
2.2 Synthesis of D-glucose grafted graphene (Glu-G)
GO was synthesized by a modified Hummers Method reported previously.27 The synthesis of the Glu-G followed an esterification procedure in the presence of DMAP and DCC as follows: purified GO (50 mg) was dissolved in dimethyl sulfoxide (DMSO, 20 mL) by sonication for 1 h and gently stirred for another 30 min. DCC (2.0 g, 9 mmol) and DMAP (0.15 g, 1.3 mmol) were then added gradually into the solution. After another 30 min of stirring, D-glucose (0.4 g, 2.2 mmol) dissolved in 10 mL DMSO was added to the solution and the resulting mixture was stirred at 60 °C for 4 days. Methanol (200 mL) was added to the mixture thereafter and the suspension was filtered and washed with acetone (300 mL). To completely remove the non-reacted GO and D-glucose, the residue was washed 3 times with hot water and filtered. The resulting solid, namely, Glu-GO, was dissolved in 30 mL DDW by sonication for 1 h. After 2 mL of hydrazine hydrate was added, the solution was heated to 60 °C for 10 h with stirring. Then, the suspension was filtered through a PTFE membrane and the residue was washed with methanol (3 × 20 mL) and hot water (3 × 20 mL). Finally, the Glu-G was dried in the vacuum oven at 60 °C for 24 h. The reaction scheme is shown in Fig. 1.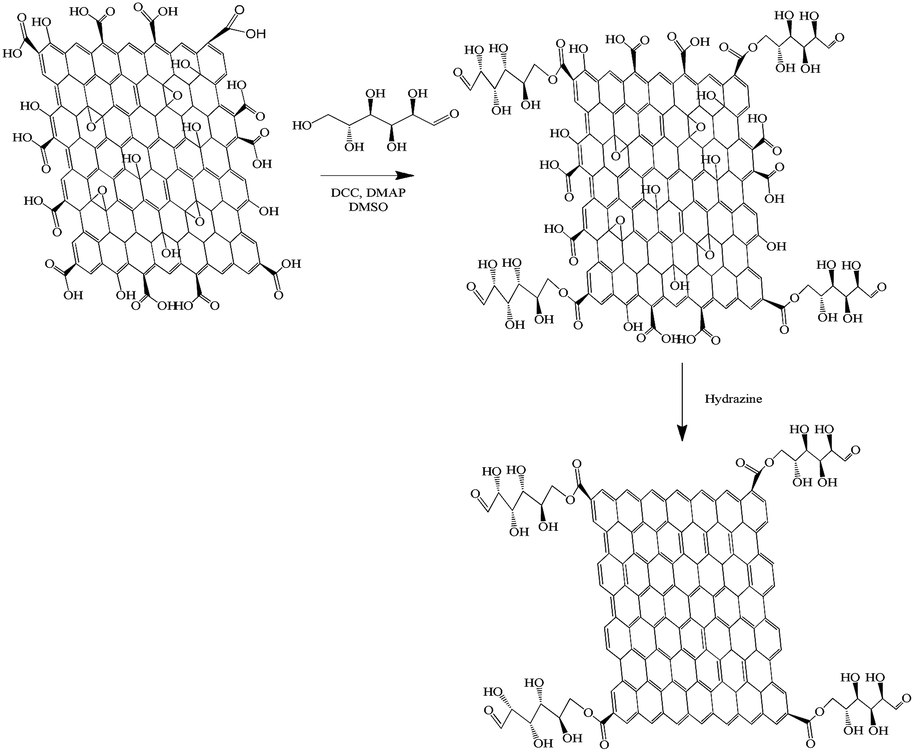 | ||
| Fig. 1 The reaction scheme for synthesizing Glu-G. | ||
2.3 Preparation of Glu-G/PVA nanocomposite
Glu-G (50 mg) was first dissolved in DDW (100 mL) and sonicated for 2 h. At the same time, PVA (0.5 g) was placed into 50 mL DDW and heated at 85 °C for 1 h with continuous stirring. After a clear PVA solution was obtained, Glu-G solution was added dropwise to the solution. After all the Glu-G solution was added, the mixture was stirred at room temperature for another 30 min. The mixture was then poured into a glass dish and kept at room temperature until a constant weight was obtained. Finally, the PVA/Glu-G 1.0 wt% film was obtained. For comparison, pure PVA film was also prepared following the similar procedure.2.4 Preparation of Glu-G/PMMA nanocomposite
Glu-G (50 mg) was first dissolved in DMF (100 mL) and sonicated for 1 h to obtain a homogeneous dispersion. At the same time, PMMA (0.5 g) was dissolved in DMF (20 mL) with continuous stirring. Thereafter, the Glu-G dispersion was added into the PMMA solution and the resulting suspension was sonicated for another 1 h. The Glu-G/PMMA suspension was then transferred to a glass dish and kept at 100 °C until a constant weight was obtained. To totally remove the residual DMF, the film was dried in a vacuum oven at 100 °C for another 24 h. Finally, the PMMA/Glu-G 1.0 wt% film was obtained. For comparison, pure PMMA film was also prepared by the similar procedure.2.5 Characterization
FTIR spectra were recorded using a Perkin Elmer 100 spectrophotometer with a resolution of 4 cm−1 and 16 scans. TGA was carried out using a Mettler Toledo TGA 1 Simultaneous Thermal analyser with the temperature increasing from 25 to 800 °C at a heating rate of 10 °C min−1. Raman spectra were performed using a Horiba Jobin Yvon HR800 Raman spectrometer, which was equipped with an Ar laser (487.97 nm, 180 mW) as the excitation light source, and an Olympus BX41 microscope. UV-vis absorption spectra were recorded from 200 to 800 nm with a Biochrom Libra S35 UV/Vis Spectrophotometer. The X-ray diffraction (XRD) was performed using Cu Kα radiation source (1.54 Å) in a Rigaku Smartlab XRD instrument. Transmission electron microscopy (TEM) images of the Glu-G were recorded using a JEOL JEM-2100F TEM instrument was operated at 200 kV. The atomic force microscopy (AFM) image of the Glu-G was investigated by a Bruker Nanoscope 8 SPM instrument using a tapping mode. Scanning electron microscopy (SEM) was conducted by the JEOL SEM 6490 to examine the fractured surface of the polymer matrices and their nanocomposites. The samples were coated with a thin layer of gold before observation. Differential scanning calorimetry (DSC) was carried out using a Perkin Elmer Pyris 1 DSC analyzer under nitrogen atmosphere. A universal tensile testing machine, Instron 5566, was used to test the mechanical properties. The testing was carried out at room temperature with a cell load of 500 N, and the gauge length was 20 mm with an extension rate of 5 mm min−1. The samples were cut into a ribbon-like shape with dimensions of 60 × 5 × 0.1 mm3 before testing.3. Results and discussion
3.1 Characterization of Glu-G
The formation of the Glu-G was characterized first by FTIR spectra shown in Fig. 2(a). It could be seen that after the GO was grafted with the D-glucose (which is Glu-GO), a new peak at around 1735 cm−1 (C![[double bond, length as m-dash]](https://www.rsc.org/images/entities/char_e001.gif) O stretching vibration) appeared and the intensity of the original peak at 1705 cm−1 became weaker, indicating that some carboxylic acid groups present in the GO had been converted to the ester groups. After the Glu-GO was reduced to Glu-G, the peak at 1705 cm−1 disappeared and that at ∼1100 cm−1 (C–O–C stretching vibration) became weaker, indicating the unreacted –COOH groups and the epoxy groups were eliminated by the hydrazine (see Fig. 1). Besides, the intensity of the two peaks at around 2900 cm−1 (–CH– stretching vibration) increased significantly, indicating that the resulting product contained more CH groups (on the glucose backbone). The peak intensity at around 3100–3500 cm−1 (–OH stretching vibration) was still relatively strong, indicating that the grafted D-glucose was not considerably affected by hydrazine.
O stretching vibration) appeared and the intensity of the original peak at 1705 cm−1 became weaker, indicating that some carboxylic acid groups present in the GO had been converted to the ester groups. After the Glu-GO was reduced to Glu-G, the peak at 1705 cm−1 disappeared and that at ∼1100 cm−1 (C–O–C stretching vibration) became weaker, indicating the unreacted –COOH groups and the epoxy groups were eliminated by the hydrazine (see Fig. 1). Besides, the intensity of the two peaks at around 2900 cm−1 (–CH– stretching vibration) increased significantly, indicating that the resulting product contained more CH groups (on the glucose backbone). The peak intensity at around 3100–3500 cm−1 (–OH stretching vibration) was still relatively strong, indicating that the grafted D-glucose was not considerably affected by hydrazine.
 | ||
| Fig. 2 (a) FTIR spectra; (b) TGA curves (under nitrogen); (c) Raman spectra and (d) XRD patterns of GO, Glu-GO and Glu-G. | ||
TGA carried out in a nitrogen atmosphere was then used to investigate the grafting rate of Glu-G, as shown in Fig. 2(b). It can be seen from the curves that the percentage weight loss was about 40% for GO and 52% for Glu-GO at 600 °C. Based on this result, it was calculated that there was about 23 wt% of glucose and 77 wt% of GO in the Glu-GO. After the Glu-GO was reduced to the Glu-G, the weight loss percentage of the Glu-G at 600 °C was about 25%. Because the oxygen containing groups had been eliminated through the reducing process,28 the weight loss of the Glu-G was mainly due to the loss of D-glucose. Because the weight content of the D-glucose obtained from the results of Glu-GO and Glu-G were similar, it was confirmed that D-glucose was grafted on the graphene and the grafting rate was ∼20%.
It is well-known that the oxidation of graphite partially destroys its sp2 structure. Thus, Raman spectroscopy was conducted to detect the structure deformation and Fig. 2(c) presents the Raman spectra of the GO, the Glu-GO and the Glu-G. It can be observed that all three materials had two characteristic peaks at around 1350 cm−1 and 1580 cm−1, which were generally named as the D band and G band, respectively. The D band indicates the carbons with sp3 hybridization, while the G band represents sp2 hybridized carbons. The intensity ratio of these two bands (ID/IG) is often introduced to speculate the degree of the structure defect. From Fig. 2(c), ID/IG for GO was calculated to be about 0.86. After the D-glucose was covalently attached to the GO, the G band red shifted to 1572 cm−1 and the ID/IG for the Glu-GO was lower (0.80) compared to that of the GO. The sp2 domains increased during the reaction process, resulting in the decrease of the ID/IG ratio. When Glu-GO converted to Glu-G after the reduction process, an increase of ID/IG (1.09) was observed, indicating that the reduction decreased the topological disorder of the graphene due to the removal of the oxygen groups.29 The peaks at around 2700 cm−1 are known as the 2D band of the carbon materials. It was also observed from Fig. 2(c) that although the peak position of the Glu-G at the 2D band shifted a little, the peak intensity of the Glu-G was almost the same as the GO, indicating that Glu-G could also be exfoliated to a few layers in the solvents.
Fig. 2(d) shows the XRD patterns of graphite, GO, Glu-GO and Glu-G. It can be seen that the graphene had a characteristic peak at ∼26° (002) with a d spacing of 0.34 nm.30 After oxidation, GO had a characteristic peak at ∼11° (001) with a much larger d spacing of 0.81 nm due to the existence of the oxygen groups. After D-glucose was grafted on the GO, the peak of the Glu-GO shifted to a lower value, indicating that the introduction of the D-glucose gave rise to the interlayer spacing of the graphene sheet. The Glu-G had a broad peak at ∼24° (002), which implied that the covalent attachment of the D-glucose did not destroy the crystalline structure of the graphene.
Fig. 3(a) and (b) show the TEM images of the Glu-G. It can be observed that the Glu-G was able to be exfoliated to very few layers. The selected area electron diffraction (SAED) pattern of the Glu-G showed well-defined diffraction spots, indicating a crystalline structure of the reduced Glu-G. Fig. 3(c) presents the UV absorption spectra of GO and Glu-G in water (with the concentration of 0.05 mg mL−1). It is well known that the absorption peak λmax represents the π conjugation of the investigated material. Compared with that of GO (∼220 nm), the λmax of Glu-G (∼280 nm) red-shifted to a higher value, indicating that the conjugation system of the Glu-G was restored after the reduction process.31
 | ||
| Fig. 3 (a) TEM and (b) high resolution TEM images of Glu-G (inset: SAED pattern); (c) UV-vis spectra of 0.05 mg mL−1 Glu-G and GO solutions; (d) digital images of graphene dispersions in (1) water and (3) DMF, and Glu-G dispersions in (2) water and (4) DMF; UV-vis spectra of Glu-G in (e) water and (f) DMF at different concentrations. The insets show the relationship between the absorbance and the concentration of the Glu-G. | ||
The introduction of the D-glucose on the surface of the graphene also affects the dispersibility of the functionalized graphene. Because the D-glucose contains a number of oxygen containing groups along its backbone, Glu-G has a much better dispersibility than GO in both aqueous and organic solvents. As shown in Fig. 3(d), Glu-G was able to be dispersed in water and DMF homogeneously without aggregation for more than 2 months. The UV-vis spectra of the Glu-G in water and DMF are shown in Fig. 3(e) and (f). From the inset figures, it can be seen that the Glu-G solution in both water and DMF obeys Beer's law. Following Beer's law, the extinction coefficient for the Glu-G in water was 36.4 mL mg−1 cm−1, with a maximal solubility of 0.58 mg mL−1, and the extinction coefficient for Glu-G in DMF was 42.2 mL mg−1 cm−1, with a maximal solubility of 0.61 mg mL−1. The good dispersion of Glu-G in both aqueous and organic solvents facilitates the fabrication of the Glu-G based nanocomposites for both aqueous soluble and organic soluble polymers.
The surface morphologies of GO and Glu-G were then investigated by AFM, as shown in Fig. 4. As can be seen from the height profile, GO had an average thickness of ∼1.0 nm, which was in accordance with the previous studies.32 Moreover, Glu-G sheet had an average thickness of ∼3.2 nm, which is much thicker than that of GO. This result was mainly ascribed to the introduction of the D-glucose on both sides of the graphene sheets.
 | ||
| Fig. 4 AFM images of GO (left) and Glu-G (right). | ||
3.2 Characterization of Glu-G/PVA and Glu-G/PMMA nanocomposites
The structures of Glu-G/PVA and Glu-G/PMMA nanocomposites were first characterized in terms of the morphology of fractured surface by SEM, as shown in Fig. 5(a) and (b). It can be clearly observed that the surfaces of both PVA/Glu-G and PMMA/Glu-G present a uniformly layered structure. In addition to the homogeneous dispersion of the Glu-G in the polymer matrices, the Glu-G sheets also tended to be distributed parallel to the direction of the polymers. Well-distributed Glu-G in the polymer matrices indicated that the Glu-G had very good bonding interactions with both PVA and PMMA. | ||
| Fig. 5 SEM images of the fractured surface of (a) PVA/Glu-G and (b) PMMA/Glu-G nanocomposites; (c) XRD patterns and (d) FTIR spectra of PVA, PMMA and their nanocomposites. | ||
To further investigate the structures of the nanocomposites, XRD analysis was conducted and the results are shown in Fig. 5(c). It was observed that pure PVA showed characteristic peaks at ∼20°, which corresponded to the crystalline phase of the PVA. The PMMA had two very broad peaks at ∼14.5° and ∼30°, revealing its amorphous structure.33 After Glu-G was incorporated, the peaks of the resulting nanocomposites were almost the same as the pristine polymers, suggesting that Glu-G sheets were fully exfoliated and well dispersed without any change the structure of the polymer matrices.
It was believed that strong interactions between the Glu-G and the polymers resulted in a uniform dispersion of the Glu-G, and the interactions were confirmed by the FTIR spectra, as shown in Fig. 5(d). Taking PVA and PVA/Glu-G as an example, it was seen that pure PVA had a wide band at ∼3455 cm−1, which was attributed to the stretching vibration of O–H bond. When Glu-G was incorporated in PVA, the peak became broader and shifted to a lower wavenumber at ∼3447 cm−1. This was due to the strong interactions between Glu-G and PVA. Because D-glucose has a number of oxygen containing groups, such as formyl and hydroxyl groups, Glu-G was able to form interactions through hydrogen bonding with –OH groups along the PVA chains, resulting in the shift of –OH in FTIR spectra. Analogously, Glu-G also formed hydrogen bonds with PMMA due to the existence of methyl ester groups within the PMMA backbone. It could be seen from the spectra that the intensity at ∼1728 cm−1 (CO stretching vibration) in PMMA/Glu-G nanocomposite increased compared to that of the pristine PMMA, which might have resulted from the interactions between the –OH groups in the Glu-G and the COOCH3 groups in the PMMA. The schematic illustration of the interactions between the filler and the matrices are shown in Fig. 6. Unlike pristine graphene, because Glu-G contains D-glucose within its body, Glu-G is more dispersible in both solvents and polymers than the pristine graphene without the introduction of foreign compatibilizers. It is thus expected that the synthesized Glu-G will be good reinforcing filler for polymers with oxygen-containing and nitrogen-containing groups such as polyurethane (PU) and resin.
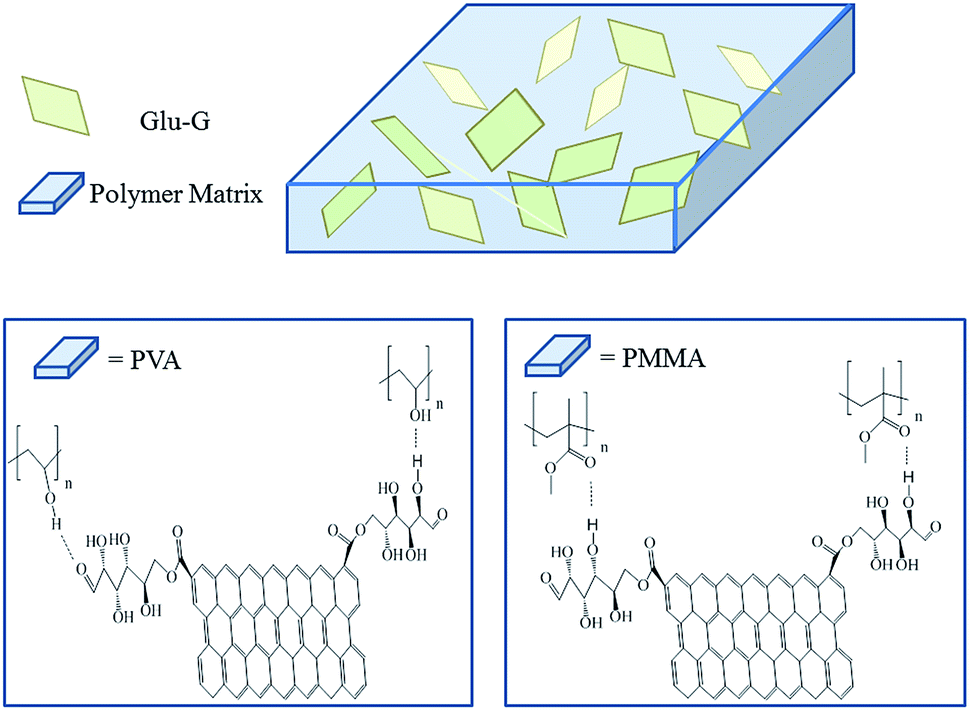 | ||
| Fig. 6 Schematic illustrations of the interactions between the filler and matrices. | ||
3.3 Thermal and mechanical properties of Glu-G/PVA and Glu-G/PMMA nanocomposites
The glass transition temperatures of PVA, PMMA and their nanocomposites were investigated by DSC and the results are shown in Fig. 7(a) and (b). Compared with that of the pure polymer, the glass transition temperature (Tg) of PVA/Glu-G nanocomposite increased from 66.8 to 69.1 °C, and the Tg of the PMMA/Glu-G nanocomposite increased from 118.7 to 120.4 °C. The increased Tg of the resulting nanocomposites was ascribed to the uniform dispersion of Glu-G in the polymer matrices and the strong hydrogen bonding between them, which was in accordance with some previous studies.34,35 The TGA measurements were carried out in an air atmosphere and the results are shown in Fig. 7(c) and (d). It was clearly seen that the nanocomposites were much more thermally stable than the pure polymers. For example, the degradation temperatures at 20% weight loss of both the PVA/Glu-G and the PMMA/Glu-G nanocomposites were higher than their pristine polymers. The improved thermal stability of the Glu-G filled nanocomposites was also ascribed to the good interactions between Glu-G and polymers. | ||
| Fig. 7 DSC curves of (a) PVA, PVA/Glu-G nanocomposite, and (b) PMMA and PMMA/Glu-G nanocomposite; and TGA curves of (c) PVA, PVA/Glu-G nanocomposite, and (d) PMMA and PMMA/Glu-G nanocomposite. | ||
It is well-known that the mechanical properties of a polymer could be influenced tremendously by the nanofillers incorporated into its matrix.36,37 Therefore, a typical stress–strain test was conducted, with the results shown in Fig. 8(a) and (b) and Table 1. It was clearly observed that the incorporation of the Glu-G notably improved the mechanical strength of the PVA and PMMA. Although the elongation of the nanocomposites decreased slightly, the Young's modulus and the tensile stress of both the PVA and PMMA nanocomposites improved significantly compared to those of the pristine polymers (Table 1). The incorporation of the Glu-G decreased the crystallinity of the polymers. The decrease in polymer crystallinity made the prepared nanocomposites tougher and easier to be broken down, which caused a slight decrease in the elongation of the nanocomposites. However, the strong hydrogen bonding interactions between the Glu-G and the polymers facilitated an effective load transfer from the polymer matrices to the filler when the nanocomposites were under stretching force, resulting in the improvement in the Young's modulus and the tensile stress.
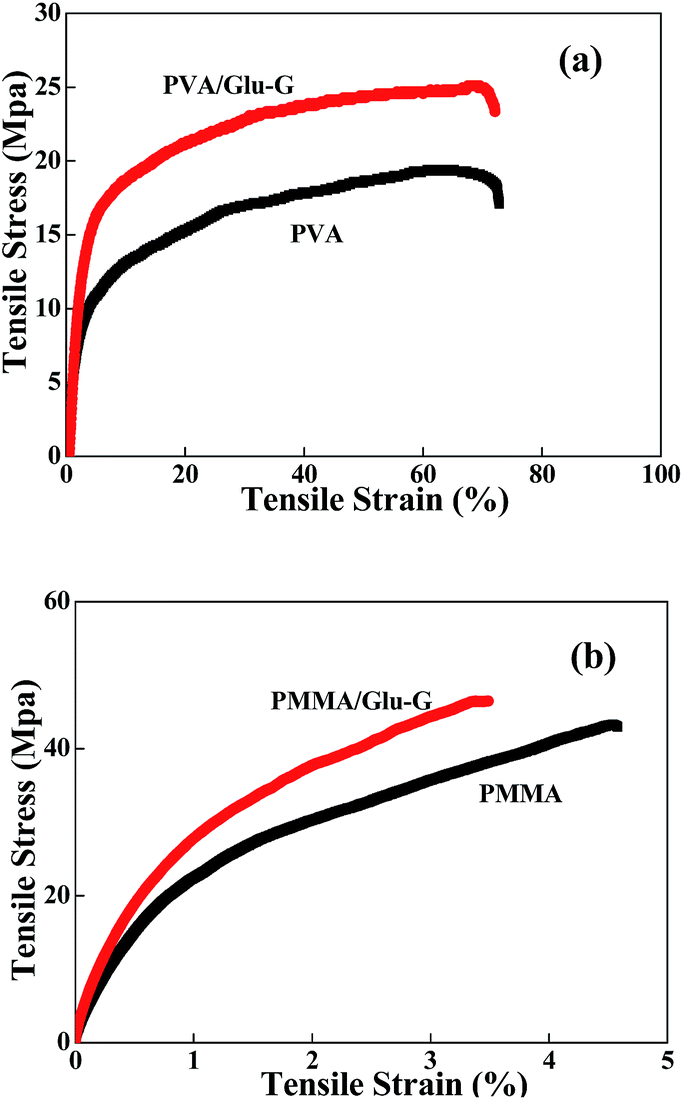 | ||
| Fig. 8 Stress–strain curves of (a) PVA, PVA/Glu-G nanocomposite, and (b) PMMA and PMMA/Glu-G nanocomposite. | ||
| Samples | Tensile stress (MPa) | Tensile strain (%) | Modulus (MPa) |
|---|---|---|---|
| PVA | 18.4 | 71.7 | 526 |
| PVA/Glu-G 1.0 wt% | 25.1 | 70.3 | 808 |
| PMMA | 43.1 | 4.6 | 14.9 |
| PVA/Glu-G 1.0 wt% | 46.6 | 3.5 | 21.2 |
4. Conclusions
In conclusion, D-glucose was covalently attached to graphene and the grafting rate was about 20%. The functionalized graphene had good dispersibility in both the aqueous and organic solvents, namely, water and DMF. The introduction of the D-glucose also improved the affinity of the functionalized graphene for the polymers PVA and PMMA. Based on the XRD and SEM results, it was found that the Glu-G was homogeneously dispersed in both the PVA and PMMA matrices and Glu-G sheets tended to align parallel to the direction of the polymers. It was then found that the Glu-G interacted with the polymers through hydrogen bonding. The strong interactions between the filler and the matrices also improved the mechanical and thermal properties of the polymers, which were validated by DSC, TGA and mechanical testing. The results suggest that the synthesized Glu-G is a desirable reinforcing nanofiller for polymers with oxygen-containing and nitrogen-containing groups. The interactions between the Glu-G and the polymers could effectively improve the thermal and mechanical properties of the matrix polymers. Compared with those CFGs, which could only be dispersed in either aqueous or organic solvents, the Glu-G synthesized in this study has wider application potential for more polymer matrices.Acknowledgements
The work was supported by the General Research Fund (Project no. 532712) of the Research Grants Council of Hong Kong and The Hong Kong Polytechnic University scholarship.Notes and references
- W. Zeng, X. Tao, S. Chen, S. Shang, H. Chan and A. S. Choy, Energy Environ. Sci., 2013, 6, 2631–2638 CAS.
- Y. Xu, S. M. Shang and J. Huang, Polym. Test., 2010, 29, 1007–1013 CrossRef CAS PubMed.
- S. M. Shang, L. Gan and M. C. W. Yuen, Compos. Sci. Technol., 2013, 86, 129–134 CrossRef CAS PubMed.
- S. M. Shang, W. Zeng and X. M. Tao, Sens. Actuators, B, 2012, 166, 330–337 CrossRef PubMed.
- Q. Zhang, J. Pan, X. Yi, L. Li and S. M. Shang, Org. Electron., 2012, 13, 1289–1295 CrossRef CAS PubMed.
- Y. Zeng, L. Qiu, K. Wang, J. Yao, D. Li, G. P. Simon, R. Wang and H. Wang, RSC Adv., 2013, 3, 887–894 RSC.
- R. S. Diggikar, D. J. Late and B. B. Kale, RSC Adv., 2014, 4, 22551–22560 RSC.
- J. Zhao, L. Zhang, H. Xue, Z. Wang and H. Hu, RSC Adv., 2012, 2, 9651–9659 RSC.
- S. M. Shang, L. Gan, M. C. W. Yuen, S. X. Jiang and N. M. Luo, Composites, Part A, 2014, 66, 135–141 CrossRef CAS PubMed.
- W. Ai, J.-Q. Liu, Z.-Z. Du, X.-X. Liu, J.-Z. Shang, M.-D. Yi, L.-H. Xie, J.-J. Zhang, H.-F. Lin, T. Yu and W. Huang, RSC Adv., 2013, 3, 45–49 RSC.
- J. Guo, L. Ren, R. Wang, C. Zhang, Y. Yang and T. Liu, Composites, Part B, 2011, 42, 2130–2135 CrossRef PubMed.
- J. Wang, J. Wu, W. Xu, Q. Zhang and Q. Fu, Compos. Sci. Technol., 2014, 91, 1–7 CrossRef CAS PubMed.
- N. Y. Yuan, F. F. Ma, Y. Fan, Y. B. Liu and J. N. Ding, Composites, Part A, 2012, 43, 2183–2188 CrossRef CAS PubMed.
- O. J. Yoon, C. Y. Jung, I. Y. Sohn, H. J. Kim, B. Hong, M. S. Jhon and N.-E. Lee, Composites, Part A, 2011, 42, 1978–1984 CrossRef PubMed.
- D. Spasevska, V. Daniloska, G. P. Leal, J. B. Gilev and R. Tomovska, RSC Adv., 2014, 4, 24477–24483 RSC.
- D. W. Boukhvalov, RSC Adv., 2013, 3, 7150–7159 RSC.
- B. Shen, W. Zhai, M. Tao, D. Lu and W. Zheng, Compos. Sci. Technol., 2013, 77, 87–94 CrossRef CAS PubMed.
- Y. Mo, M. Yang, Z. Lu and F. Huang, Composites, Part A, 2013, 54, 153–158 CrossRef CAS PubMed.
- B. Shen, W. Zhai, D. Lu, J. Wang and W. Zheng, RSC Adv., 2012, 2, 4713–4719 RSC.
- O.-K. Park, S.-G. Kim, N.-H. You, B.-C. Ku, D. Hui and J. H. Lee, Composites, Part B, 2014, 56, 365–371 CrossRef CAS PubMed.
- C. Deetuam, C. Samthong, S. Thongyai, P. Praserthdam and A. Somwangthanaroj, Compos. Sci. Technol., 2014, 93, 1–8 CrossRef CAS PubMed.
- K. Liu, S. Chen, Y. Luo, D. Jia, H. Gao, G. Hu and L. Liu, Compos. Sci. Technol., 2013, 88, 84–91 CrossRef CAS PubMed.
- W. Yu, J. Fu, X. Dong, L. Chen and L. Shi, Compos. Sci. Technol., 2014, 92, 112–119 CrossRef CAS PubMed.
- E. Ou, Y. Xie, C. Peng, Y. Song, H. Peng, Y. Xiong and W. Xu, RSC Adv., 2013, 3, 9490–9499 RSC.
- C. T. Wass and W. L. Lanier, Mayo Clin. Proc., 1996, 71, 801–812 CrossRef CAS.
- K. P. Loh, Q. Bao, P. K. Ang and J. Yang, J. Mater. Chem., 2010, 20, 2277–2289 RSC.
- L. Y. Li, K. Q. Xia, L. Li, S. M. Shang, Q. Z. Guo and G. P. Yan, J. Nanopart. Res., 2012, 14, 908 CrossRef.
- S. Chandrasekaran, G. Faiella, L. A. S. A. Prado, F. Tölle, R. Mülhaupt and K. Schulte, Composites, Part A, 2013, 49, 51–57 CrossRef CAS PubMed.
- T. Kuila, S. Bose, P. Khanra, N. H. Kim, K. Y. Rhee and J. H. Lee, Composites, Part A, 2011, 42, 1856–1861 CrossRef PubMed.
- H. Yang, C. Shan, F. Li, D. Han, Q. Zhang and L. Niu, Chem. Commun., 2009, 26, 3880–3882 RSC.
- D. Li, M. B. Mueller, S. Gilje, R. B. Kaner and G. G. Wallace, Nat. Nanotechnol., 2008, 3, 101–105 CrossRef CAS PubMed.
- C. Xu, J. Gao, H. Xiu, X. Li, J. Zhang, F. Luo, Q. Zhang, F. Chen and Q. Fu, Composites, Part A, 2013, 53, 24–33 CrossRef CAS PubMed.
- Y. Li, B. Zhang and X. Pan, Compos. Sci. Technol., 2008, 68, 1954–1961 CrossRef CAS PubMed.
- X. M. Yang, L. Li, S. M. Shang and X. M. Tao, Polymer, 2010, 51, 3431–3435 CrossRef CAS PubMed.
- J. Shen, B. Yan, T. Li, Y. Long, N. Li and M. Ye, Composites, Part A, 2012, 43, 1476–1481 CrossRef CAS PubMed.
- S. Biswas, H. Fukushima and L. T. Drzal, Composites, Part A, 2011, 42, 371–375 CrossRef PubMed.
- S. C. Shiu and J. L. Tsai, Composites, Part B, 2014, 56, 691–697 CrossRef CAS PubMed.
| This journal is © The Royal Society of Chemistry 2015 |