DOI:
10.1039/C4RA17334A
(Paper)
RSC Adv., 2015,
5, 18252-18257
Spontaneous resolution of P and M helical copper(II) MOFs built from achiral precursors†
Received
31st December 2014
, Accepted 21st January 2015
First published on 21st January 2015
Abstract
Two enantiomorphic (P and M) helical coordination polymers of [Cu(μ1,3-N3)(L)]n {HL = 2-(dimethylamino)ethyliminomethylnaphthalen-2-ol} have been synthesized by the self-assembly of the achiral precursors via spontaneous resolution. The structures have been confirmed by single crystal X-ray crystallography. Solid-state circular dichroism spectra have confirmed the chirality of both P and M helical chains.
Introduction
A rich diversity of architectures of coordination polymers could be produced by linking transition metal centers with bridging ligands.1 These coordination polymers could have potential applications in molecular-based ferromagnets, non-linear optics and ferroelectrics.2 Among them, the helical coordination polymers constitute a special class and deserve a special mention because of their potential applications in materials science, chemical sensing, chiral magnets, enantio-selective catalysis, non-linear optical materials and biomimetic chemistry.3 A variety of bridging groups have been employed for the synthesis of coordination polymers; among them pseudo-halide bridged complexes have attracted the attention of coordination chemists for their versatile modes of binding metal centres.4 Several N2O donor Schiff bases are popularly used as blocking ligands in preparing such complexes.5 Focusing on copper(II), pseudo-halide bridged complexes have received considerable attention because of their potential application in bioinorganic modeling chemistry,6 magnetic materials7 and catalysis.8 On the other hand, Great effort has been made to design and construct coordination polymers with single-, double- or multi-strand helical structures by several groups.9 The helical structures induce chirality, but due to the presence of both P (right-handed) and M (left handed) helices, they are usually optically inactive.10 However, if complexes belong to chiral space groups, either a P or M helix is present in it and consequently, the complex is chiral as well as optically active. The most effective method for the synthesis of a chiral helix is to use a chiral molecule as the primary linker or as an auxiliary ligand.11 However, due to high cost of chiral ligands, it is often highly desirable to create chiral compounds from achiral precursors by spontaneous chiral resolution.12
Flack parameter13 usually gives a rough idea about the absolute configuration (the left- or right-handedness) for any chiral solid. A value close to 0 suggests correct absolute configuration of a crystal. If the crystal is twinned or racemic, the value goes to ~0.5. On the other hand, if the value is near 1, then it could be concluded that inverted structure is the correct absolute configuration of that crystal.
Our attempts with a N2O donor Schiff base, 2-(dimethylamino)ethyliminomethylnaphthalen-2-ol (HL) afforded copper(II) helices, [Cu(μ1,3-N3)(L)]n, with end-to-end azide bridges. Single crystal X-ray data, values of Flack parameter and solid state CD spectra support the presence of both P and M helices in the bulk material leading to the formation of racemic conglomerate. Herein, we report the synthesis, spectroscopic characterization and crystal structures of these P and M helical chains obtained by the self-assembly of achiral precursors via spontaneous resolution.
Experimental details
All materials were commercially available, reagent grade and used as purchased from Sigma-Aldrich without further purification. Although no problems were encountered in this work, organic ligands containing azides are potentially explosive. Only a small amount of the material should be prepared and they should be handled with care.
Synthesis of the complex
A methanol solution of N,N-dimethyl-1,2-diaminoethane (0.10 mL, 1 mmol) and 2-hydroxy-1-naphthaldehyde (172 mg, 1 mmol) was refluxed for ca. 1 h to form the tridentate Schiff base ligand HL. The ligand was not isolated. A methanol (10 mL) solution of copper(II) acetate monohydrate (200 mg, 1 mmol) was added into the methanol solution of the ligand HL to get a dark blue solution. A methanol–water solution of sodium azide (65 mg, 1 mmol) was added into the reaction mixture with constant stirring. The stirring was continued for additional 2 h. Dark blue single crystals, suitable for X-ray diffraction, were obtained after few days by slow evaporation of the solution in open atmosphere.
(Yield: 274 mg, 79%) anal. calcd for C15H17CuN5O (346.89): C, 51.94; H, 4.94; N, 20.19%. Found: C, 51.7; H, 4.8; N, 20.4%. FT-IR (KBr, cm−1): 1622 (C![[double bond, length as m-dash]](https://www.rsc.org/images/entities/char_e001.gif) N); 2067 (N3). λmax (nm) [εmax (Lit mol−1 cm−1)] (acetonitrile): 298 (3.96 × 104); 383 (2.23 × 104); 584 (3.05 × 102).
N); 2067 (N3). λmax (nm) [εmax (Lit mol−1 cm−1)] (acetonitrile): 298 (3.96 × 104); 383 (2.23 × 104); 584 (3.05 × 102).
X-ray crystallography
Eleven suitable crystals of the complex were used for data collection using a ‘Bruker SMART APEX II’ diffractometer equipped with graphite-monochromated Mo-Kα radiation (λ = 0.71073 Å). The molecular structures were solved by direct method and refined by full-matrix least squares on F2 using the SHELX-97 package.14 Non-hydrogen atoms were refined with anisotropic thermal parameters. The hydrogen atoms were placed in their geometrically idealized positions and constrained to ride on their parent atoms. Multi-scan empirical absorption corrections were applied to the data using the program SADABS.15 Details of crystallographic data and refinements of one M and one P helical MOFs are given in Table 1. CCDC reference numbers are 999066 and 1039159 respectively. Data for other M and P helices are gathered in Table S1; ESI.†
Table 1 Crystallographic data and refinement details of one M and one P helical MOFs
| Complex |
M |
P |
| Formula |
C15H17CuN5O |
C15H17CuN5O |
| Formula weight |
346.89 |
346.89 |
| Temperature (K) |
293 |
293 |
| Crystal system |
Monoclinic |
Monoclinic |
| Space group |
P21 |
P21 |
| a (Å) |
8.011(5) |
8.0058(3) |
| b (Å) |
6.852(5) |
6.8434(2) |
| c (Å) |
14.033(5) |
14.0474(5) |
| β (deg) |
103.777(5) |
103.658(2) |
| Z |
2 |
2 |
| Dcalcd (g cm−3) |
1.540 |
1.541 |
| μ (mm−1) |
1.469 |
1.469 |
| F(000) |
358 |
358 |
| Flack parameter |
0.06(6) |
0.033(14) |
| Total reflections |
2717 |
10![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) 888 888 |
| Unique reflections |
1548 |
2842 |
| Observed data [I > 2σ(I)] |
1487 |
2552 |
| No. of parameters |
157 |
199 |
| R (int) |
0.063 |
0.039 |
| R1, wR2 (all data) |
0.0883, 0.2097 |
0.0366, 0.0642 |
| R1, wR2 [I > 2σ(I)] |
0.0759, 0.1880 |
0.0309, 0.0623 |
Details of instrumentation
Elemental analyses (carbon, hydrogen and nitrogen) were performed using a PerkinElmer 240C elemental analyzer. IR spectra in KBr (4500–500 cm−1) were recorded with a PerkinElmer Spectrum Two spectrophotometer. Electronic spectra in acetonitrile were recorded on a PerkinElmer Lambda 35 UV-visible spectrophotometer. Solid state (KBr pellets) circular dichroism (CD) spectrum was recorded on a JASCO J-810 spectropolarimeter. Powder X-ray diffraction was performed on a Bruker D8 instrument with Cu-Kα radiation. Steady state photoluminescence spectra in acetonitrile were obtained in Shimadzu RF-5301PC spectrofluorometer at room temperature. Time dependent photoluminescence measurements were done using Hamamatsu MCP photomultiplier (R3809) and were analyzed by using IBHDAS6 software.
The emission of the complex is tentatively attributed to the intra ligand transitions modified by metal coordination. Intensity decay profile was fitted to the sum of exponentials series  , where αi was a factor representing the fractional contribution to the time-resolved decay of the component with a lifetime of τi. Bi-exponential function was used to fit the decay profile for the complex, with obtaining χ2 close to 1. The intensity-averaged life time (τav) was determined from the result of the exponential model using
, where αi was a factor representing the fractional contribution to the time-resolved decay of the component with a lifetime of τi. Bi-exponential function was used to fit the decay profile for the complex, with obtaining χ2 close to 1. The intensity-averaged life time (τav) was determined from the result of the exponential model using 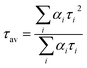 , where αi and τi are the pre-exponential factors and excited-state luminescence decay time associated with the i-th component, respectively.
, where αi and τi are the pre-exponential factors and excited-state luminescence decay time associated with the i-th component, respectively.
Result and discussion
Synthesis
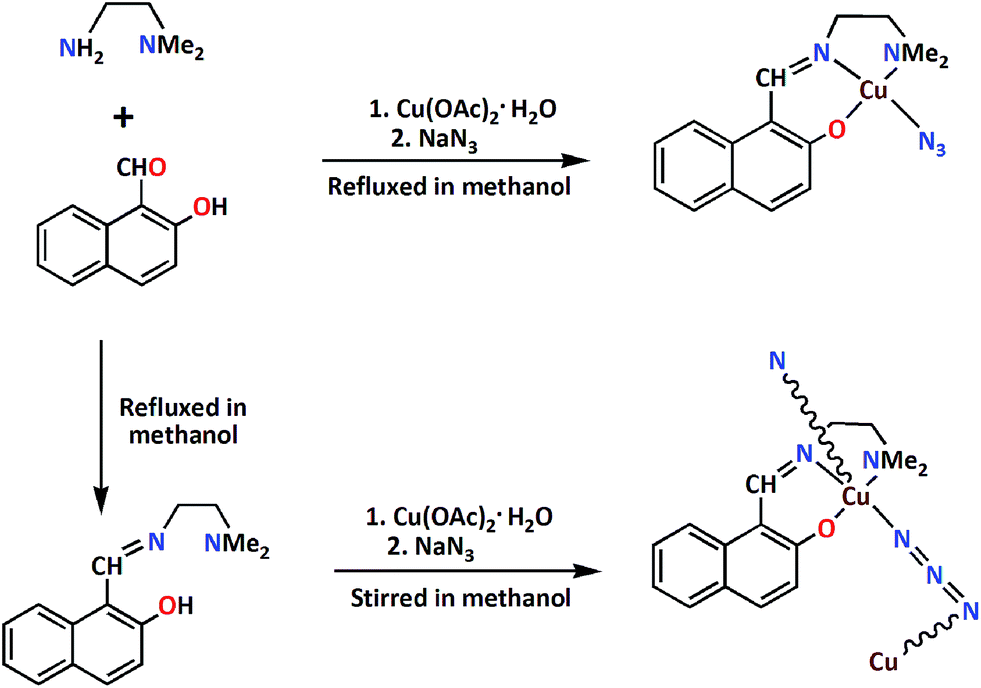
The tridentate Schiff base ligand, HL, was prepared by the 1:1 condensation of N,N-dimethyl-1,2-diaminoethane with 2-hydroxy-1-naphthaldehyde in methanol following the literature method.1f The methanol solution of HL was then made to react with copper(II) acetate monohydrate along with sodium azide to prepare M and P helices of [Cu(μ1,3-N3)(L)]n. Eleven crystals from the same reaction were randomly picked and characterized by single-crystal X-ray diffraction analyses, which showed that the bulk material contained single crystals of both M and its enantiomorph P helices almost in equal proportions. Each single crystal crystallizes in the chiral monoclinic space group P21. The Flack parameters of eleven crystals are all close to zero (Table 1 and S1; ESI†). The results demonstrate that each individual crystal is homochiral while the bulk material is a racemic conglomerate consisting of more or less 50% P and 50% M helices. It is to be noted here that a mononuclear complex, [Cu(N3)(L)], could be crystallized from an unstirred solution. The structure of this complex was reported by a different group.16 The formation of both complexes is shown in Scheme 1.
 |
| | Scheme 1 Synthetic route to the helical coordination polymer and its mononuclear analogue. | |
Structure description
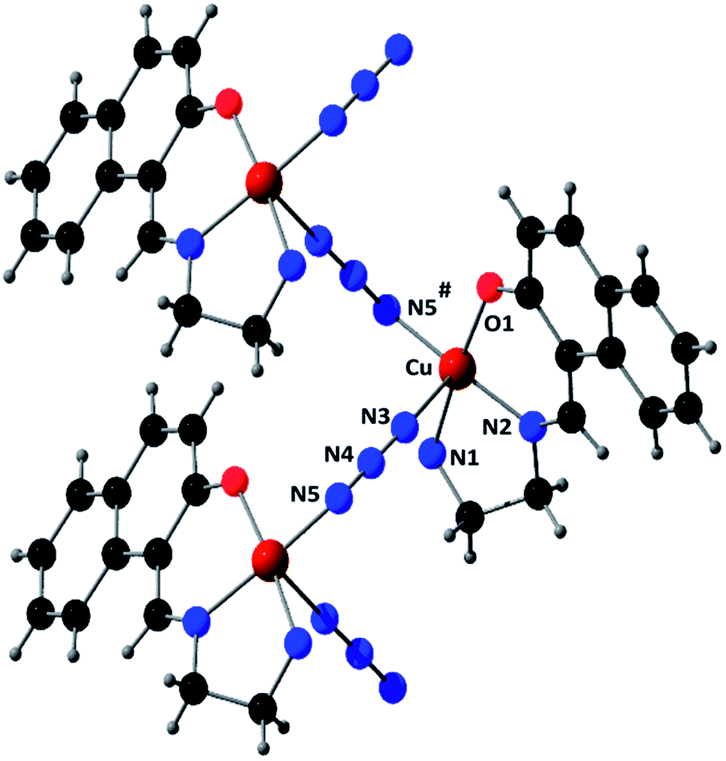
The fundamental building units for both M and P helices consist of a copper(II) centre, one deprotonated Schiff base ligand, (L)−, and an azide ion. Taking one M helix as an example, a perspective view of the complex with the selective atom-numbering scheme is shown in Fig. 1. Copper(II) centres are bridged singly by end-to-end (EE) azides with the formation of the helix. Each copper(II) centre is coordinated equatorially by one amine nitrogen atom, N(1), one imine nitrogen atom, N(2), and one oxygen atom, O(1), of the tridentate deprotonated Schiff base, (L)−, and a nitrogen atom, N(3), of the EE bridged azide; this defines the basal plane. An nitrogen atom, N(5)*, from a symmetry related (−x, −1/2 + y, 1 − z) bridging azide coordinates axially at a rather long distance furnishing an elongated square-pyramidal (4 + 1) geometry for each copper(II) center. The value of addison parameter (τ)17 is 0.047, which also suggests a slightly distorted square pyramidal geometry. In the equatorial plane, the Cu–Nimine distance {1.933(13) Å} is shorter than the Cu–Namine {2.032(13) Å} distance, as was also observed in similar systems.18 The deviations of the coordinating atoms N(1), N(2), O(1) and N(3) from the least square mean plane through them are −0.001(13), 0.047(13), 0.002(11) and 0.047(13) Å respectively. As usual for a square pyramid structure, the copper(II) is slightly pulled out of this mean square plane towards the apical donor atom at a distance 0.0944(16) Å. The closest conformation of the five-membered chelate ring, Cu(1)–N(1)–C(3)–C(4)–N(2), is envelope with puckering parameters q(2) = 0.430(15) Å and ϕ(2) = 71.8(16)°.19 The bridging azides are quasi-linear; the N–N–N angle is 174.3(16)°. The shortest Cu⋯Cu distance is 5.838(3) Å in the helix.
 |
| | Fig. 1 Perspective view of M helix with selective atom numbering scheme. Methyl groups of the amine nitrogen atoms have been omitted for clarity. Symmetry transformation: * = −x, −1/2 + y, 1 − z. | |
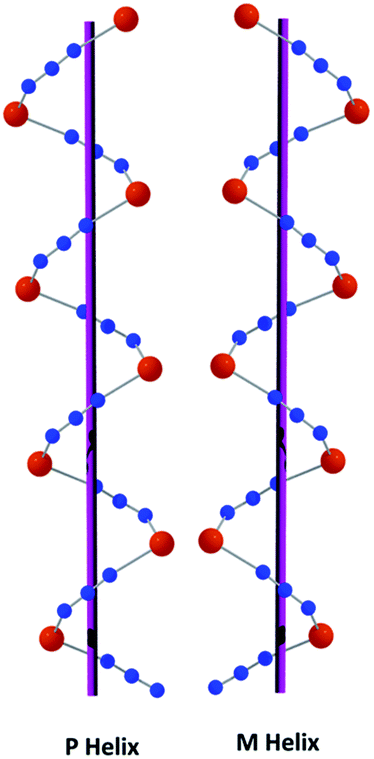
On the other hand, the P helix has a very similar structure (Fig. 2) in which each square-pyramidal (addison parameter = 0.034) copper(II) centre is coordinated equatorially by one amine nitrogen atom, N(1), one imine nitrogen atom, N(2), and one oxygen atom, O(1), of the tridentate deprotonated Schiff base, (L)−, and a nitrogen atom, N(3), of the EE bridged azide; and axially by an nitrogen atom, N(5)#, from a symmetry related (1 − x, 1/2 + y, 2 − z) end-to-end bridging azide. Cu–Nimine distance {1.939(2) Å} is shorter than the Cu–Namine {2.042(2) Å} distance, as was also observed in M helical chain. The deviations of the coordinating atoms N(1), N(2), O(1) and N(3) from the least square mean plane through them are −0.018(2), 0.019(2), −0.019(2) and 0.018(2) Å respectively and that of copper(II) from the same plane −0.1362(3) Å. The closest conformation of the five-membered chelate ring, Cu(1)–N(1)–C(3)–C(4)–N(2), is envelope with puckering parameters q(2) = 0.427(3) Å and ϕ(2) = 250.1(3)°.19 The N–N–N angle in the bridging azide is 175.4(6)°. The shortest intra-helix Cu⋯Cu distance is 5.8362(5) Å. Selected bond lengths and angles of both M and P helices are gathered in Table 2. A closer look at the both P and M helical chains are shown in Fig. 3.
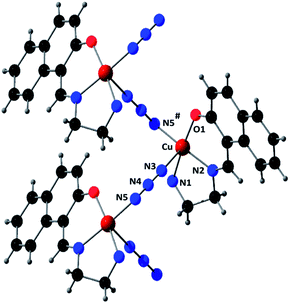 |
| | Fig. 2 Perspective view of P helix with selective atom numbering scheme. Methyl groups of the amine nitrogen atoms have been omitted for clarity. Symmetry transformation: # = 1 − x, 1/2 + y, 2 − z. | |
 |
| | Fig. 3 A closer look at the P and M helical chains of the complex. | |
Table 2 Selected bond lengths (Å) and bond angles (°) in M and P helices of the complexa
| |
M |
P |
| Symmetry transformations: * = −x, −1/2 + y, 1 − z (for M) and # = 1 − x, 1/2 + y, 2 − z (for P). |
| Cu(1)–O(1) |
1.936(10) |
1.914(2) |
| Cu(1)–N(1) |
2.032(13) |
2.042(2) |
| Cu(1)–N(2) |
1.933(13) |
1.939(2) |
| Cu(1)–N(3) |
1.945(13) |
1.962(3) |
| Cu(1)–N(5)#/* |
2.495(13) |
2.471(4) |
| O(1)–Cu(1)–N(1) |
173.7(5) |
172.36(10) |
| O(1)–Cu(1)–N(2) |
90.9(5) |
90.89(10) |
| O(1)–Cu(1)–N(3) |
92.3(5) |
91.80(11) |
| O(1)–Cu(1)–N(5)#/* |
92.9(4) |
92.85(12) |
| N(1)–Cu(1)–N(2) |
85.6(5) |
85.15(10) |
| N(1)–Cu(1)–N(3) |
90.4(5) |
91.09(11) |
| N(1)–Cu(1)–N(5)#/* |
92.2(5) |
93.50(12) |
| N(2)–Cu(1)–N(3) |
170.9(5) |
170.34(12) |
| N(2)–Cu(1)–N(5)#/* |
87.9(5) |
87.43(12) |
| N(3)–Cu(1)–N(5)#/* |
100.5(5) |
101.70(13) |
The hydrogen atom H(1A), attached with a methyl carbon, C(1), in the M helix is involved in C–H⋯π interaction with a symmetry related (1 + x, y, z) phenyl ring [C(7)–C(8)–C(9)–C(10)–C(11)–C(12)] from a neighbouring helix to form supra-molecular two-dimensional sheet as shown in Fig. 4. C–H⋯π interactions in the P helix also form similar supra-molecular two-dimensional sheet.
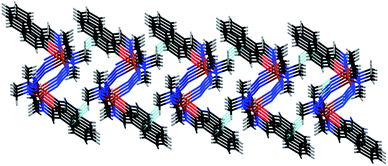 |
| | Fig. 4 Perspective view of the two dimensional supra-molecular network formed via C–H⋯π interactions in the M helix. | |
CD spectroscopy
Additional evidence for the occurrence of spontaneous resolution comes from CD spectroscopy performed on the single crystals obtained in the same crystallization. Each and every single crystal of the complex that was used for single crystal X-ray diffraction analysis, are also used for collection of CD spectrum. The results gave the expected opposite Cotton effects, positive or negative (at 298, 383 and 584 nm), revealing the formation of either M or P helices in the bulk sample under spontaneous resolution (Fig. 5).
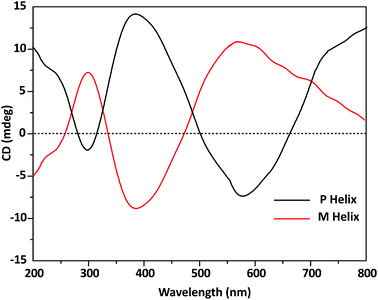 |
| | Fig. 5 Solid state (KBr pellet) CD spectra of P and M helices showing the opposite cotton effects. | |
IR, UV-Vis, fluorescence spectroscopy and PXRD
In the IR spectrum of the complex, distinct band due to the azomethine (CN) group at 1622 cm−1 (Fig. 6) is routinely noticed.1f The appearance of a strong band at 2067 cm−1 indicates the presence of μ1,3-bridged azide in the complex.20 The UV-Vis absorption spectrum (Fig. 7) of the complex in acetonitrile shows a d–d transition band at 584 nm, as expected for a square pyramidal copper(II) environment with N2O donor tridentate Schiff bases along with nitrogen donor monodentate ligands.21a The intense absorption bands below 400 nm may be assigned to ligand to metal ion charge transfer bands (LMCT).21a On the other hand, the absorption spectrum of its mononuclear analogue, [Cu(N3)(L)], shows only one d–d transition band at 604 nm along with the intense absorption bands below 400 nm due to LMCT transitions in acetonitrile solution. Although both square planar and square pyramidal complexes exhibit single symmetric bands due to the dz2 → dx2−y2 transitions, the positions of the absorption maxima should differ.21b Therefore, the difference in the value of the λmax of the mononuclear complex containing square planar copper(II) and the helical polynuclear one with square pyramidal copper(II) indicates their independent existence in solution.
 |
| | Fig. 6 IR spectrum (KBr pellet) of the bulk material. | |
 |
| | Fig. 7 UV-Vis spectrum of the bulk material in acetonitrile solution. | |
The complex exhibits luminescence in acetonitrile medium at 425 nm on exciting at 380 nm and may be assigned to intra-ligand (π–π*) fluorescence.21 The mean lifetime (τav) of the exited state is 2.72 ns at room temperature and the lifetime decay profile is shown in Fig. S1; ESI.† The experimental P-XRD pattern of the bulk product is in good agreement with the simulated XRD pattern from single-crystal X-ray diffraction, indicating consistency of the bulk sample (Fig. S2; ESI†). The simulated patterns of the complexes are calculated from the single crystal structural data (cif files) using the CCDC Mercury software.
Conclusion
In the present work, an achiral tridentate N2O donor Schiff base was used to prepare new helical chains of a copper(II) MOF via spontaneous chiral resolution. Solid state CD spectra and single crystal X-ray diffraction studies show that each individual crystal is homochiral while the bulk material is a racemic conglomerate containing both P and M helices.
Acknowledgements
This work was supported by the DST, India under FAST Track Scheme (Order no. SR/FT/CS-118/2010, dated 15/02/2012). A. B. thanks the UGC, India, for awarding a Junior Research Fellowship. Crystallographic data were collected at the DST-FIST, India funded Single Crystal Diffractometer Facility at the Department of Chemistry, Jadavpur University.
Notes and references
-
(a) C. Biswas, S. Chattopadhyay, M. G. B. Drew and A. Ghosh, Polyhedron, 2007, 26, 4411–4418 CrossRef CAS PubMed;
(b) H. Y. Zang, Y. Q. Lan, G. S. Yang, X. L. Wang, K. Z. Shao, G. J. Xu and Z. M. Su, CrystEngComm, 2010, 12, 434–445 RSC;
(c) M. Das, S. Chatterjee and S. Chattopadhyay, Polyhedron, 2014, 68, 205–211 CrossRef CAS PubMed;
(d) Z. N. Chen, H. X. Zhang, K. B. Yu, K. C. Zheng, H. Cai and B. S. Kang, J. Chem. Soc., Dalton Trans., 1998, 1133–1136 RSC;
(e) A. Bacchi, M. Carcelli, T. Chiodo and P. Pelagatti, CrystEngComm, 2010, 12, 4226–4230 RSC;
(f) M. Das and S. Chattopadhyay, Polyhedron, 2013, 50, 443–451 CrossRef CAS PubMed.
-
(a) R. Biswas, Y. Ida, M. L. Baker, S. Biswas, P. Kar, H. Nojiri, T. Ishida and A. Ghosh, Chem.–Eur. J., 2013, 19, 3943–3953 CrossRef CAS PubMed;
(b) L.-Z. Chen, D.-D. Huang, J.-Z. Ge and F.-M. Wang, Inorg. Chim. Acta, 2013, 406, 95–99 CrossRef CAS PubMed;
(c) Y. Wang, Y.-X. Che and J.-M. Zheng, Inorg. Chem. Commun., 2012, 21, 69–71 CrossRef PubMed.
-
(a) J. M. Lehn, Supra-molecular chemistry: concepts and perspectives, VCH, New York, 1995 Search PubMed;
(b) C. R. Woods, M. Benaglia, F. Cozzi and J. S. Siegel, Angew. Chem., Int. Ed., 1996, 35, 1830–1833 CrossRef CAS;
(c) C. Piguet, G. Bernardinelli and G. Hopfgartner, Chem. Rev., 1997, 97, 2005–2062 CrossRef CAS PubMed;
(d) C. Chen and K. S. Suslick, Coord. Chem. Rev., 1993, 128, 293–322 CrossRef CAS;
(e) A. Zingiryan, J. Zhang and X. Bu, Inorg. Chem., 2008, 47, 8607–8609 CrossRef CAS PubMed;
(f) E. V. Anokhina, Y. B. Go, Y. Lee, T. Vogt and A. J. Jacobson, J. Am. Chem. Soc., 2006, 128, 9957–9962 CrossRef CAS PubMed;
(g) R. Vaidhyanathan, D. Bradshaw, J.-N. Rebilly, J. P. Barrio, J. A. Gould, N. G. Berry and M. J. Rosseinsky, Angew. Chem., Int. Ed., 2006, 45, 6495–6499 CrossRef CAS PubMed;
(h) Q.-L. Zhu, T.-L. Sheng, R.-B. Fu, C.-H. Tan, S.-M. Hu and X.-T. Wu, Chem. Commun., 2010, 46, 9001–9003 RSC;
(i) Q.-C. Zhang, X.-H. Bu, Z.-E. Lin, M. Biasini, W. P. Beyermann and P.-Y. Feng, Inorg. Chem., 2007, 46, 7262–7264 CrossRef CAS PubMed;
(j) H. C. Aspinall, Chem. Rev., 2002, 102, 1807–1850 CrossRef CAS PubMed;
(k) O. Mamula and A. Zelewsky, Coord. Chem. Rev., 2003, 242, 87–95 CrossRef CAS;
(l) J. S. Seo, D. Whang, H. Lee, S. I. Jun, J. Oh, Y. J. Jeon and K. Kim, Nature, 2000, 404, 982–986 CrossRef CAS PubMed;
(m) B. Kesanli and W. Lin, Coord. Chem. Rev., 2003, 246, 305–326 CrossRef CAS PubMed.
-
(a) P. Bhowmik, H. P. Nayek, M. Corbella, N. Aliaga-Alcalde and S. Chattopadhyay, Dalton Trans., 2011, 7916–7926 RSC;
(b) P. Bhowmik, S. Chattopadhyay, M. G. B. Drew, C. Diaz and A. Ghosh, Polyhedron, 2010, 29, 2637–2642 CrossRef CAS PubMed.
-
(a) C. Biswas, M. G. B. Drew, S. Asthana, C. Desplanches and A. Ghosh, J. Mol. Struct., 2010, 965, 39–44 CrossRef CAS PubMed;
(b) P. K. Bhowmik and S. Chattopadhyay, Inorg. Chem. Commun., 2012, 22, 14–17 CrossRef PubMed;
(c) M. Das, B. N. Ghosh, A. Valkonen, K. Rissanen and S. Chattopadhyay, Polyhedron, 2013, 60, 68–77 CrossRef CAS PubMed;
(d) P. K. Bhaumik, K. Harms and S. Chattopadhyay, Inorg. Chim. Acta, 2013, 405, 400–409 CrossRef CAS PubMed.
- E. I. Solomon, U. M. Sundaram and T. E. Machonkin, Chem. Rev., 1996, 96, 2563–2606 CrossRef CAS PubMed.
-
(a) C. Biswas, M. G. B. Drew, E. Ruiz, M. Estrader, C. Diaz and A. Ghosh, Dalton Trans., 2010, 7474–7484 RSC;
(b) C. Biswas, M. G. B. Drew, A. Figuerola, S. Gómez-Coca, E. Ruiz, V. Tangoulis and A. Ghosh, Inorg. Chim. Acta, 2010, 363, 846–854 CrossRef CAS PubMed.
- A. Biswas, L. K. Das, M. G. B. Drew, C. Diaz and A. Ghosh, Inorg. Chem., 2012, 51, 10111–10121 CrossRef CAS PubMed.
-
(a) M. Albrecht, Chem. Rev., 2001, 101, 3457–3498 CrossRef CAS PubMed;
(b) S. Kitagawa, R. Kitaura and S. Noro, Angew. Chem., Int. Ed., 2004, 43, 2334–2375 CrossRef CAS PubMed;
(c) L. Han and M. Hong, Inorg. Chem. Commun., 2005, 8, 406–419 CrossRef CAS PubMed;
(d) M. Kimura, M. Sano, T. Muto, K. Hanabusa and H. Shirai, Macromolecules, 1999, 32, 7951–7953 CrossRef CAS;
(e) W. Wei, K. Wu, J. He, W. Zheng and X. Xiao, CrystEngComm, 2011, 13, 52–54 RSC.
- P. Bhowmik, S. Chattopadhyay, M. G. B. Drew and A. Ghosh, Inorg. Chim. Acta, 2013, 395, 24–32 CrossRef CAS PubMed.
-
(a) A. Hu, H. L. Ngo and W. B. Lin, Angew. Chem., Int. Ed., 2003, 42, 6000–6003 CrossRef CAS PubMed;
(b) Y. Liu, G. Li, X. Li and Y. Cui, Angew. Chem., Int. Ed., 2007, 46, 6301–6304 CrossRef CAS PubMed;
(c) P. S. Navare and J. C. MacDonald, Cryst. Growth Des., 2011, 11, 2422–2428 CrossRef CAS;
(d) C. J. Kepert, T. J. Prior and M. J. Rosseinsky, J. Am. Chem. Soc., 2000, 122, 5158–5168 CrossRef CAS.
- W. Zheng, Y. Wei, C. Tian, X. Xiao and K. Wu, CrystEngComm, 2012, 14, 3347–3350 RSC.
- H. D. Flack and G. Bernardinelli, Chirality, 2008, 20, 681–690 CrossRef CAS PubMed.
-
(a) G. M. Sheldrick, Acta Crystallogr., 2008, A64, 112–122 CrossRef PubMed;
(b) G. M. Sheldrick, SHELXS-97 and SHELXL-97: Program for Structure Solution, University of Göttingen, Institute fur Anorganische Chemieder Universitat, Gottingen, Germany, 1997 Search PubMed.
- G. M. Sheldrick, SADABS: Software for Empirical Absorption Correction, University of Göttingen, Institute fur Anorganische Chemieder Universitat, Gottingen, Germany, 1999–2003 Search PubMed.
- Q.-Y. Zhu, Y.-J. Wei and F.-W. Wang, Acta Crystallogr., 2006, E62, m983–m985 Search PubMed.
- A. W. Addison, T. N. Rao, J. Reedjik, J. van Rinj and C. G. Verscholar, J. Chem. Soc., Dalton Trans., 1984, 1349–1356 RSC.
- M. Das and S. Chattopadhyay, J. Mol. Struct., 2013, 1051, 250–258 CrossRef CAS PubMed.
-
(a) D. Cremer and J. A. Pople, J. Am. Chem. Soc., 1975, 97, 1354–1358 CrossRef CAS;
(b) D. Cremer, Acta Crystallogr., 1984, B40, 498–500 CrossRef CAS;
(c) J. C. A. Boeyens, J. Cryst. Mol. Struct., 1978, 8, 317–320 CrossRef.
- S. Jana, P. Bhowmik, M. Das, P. P. Jana, K. Harms and S. Chattopadhyay, Polyhedron, 2012, 37, 21–26 CrossRef CAS PubMed.
-
(a) P. K. Bhaumik, K. Harms and S. Chattopadhyay, Polyhedron, 2014, 68, 346–356 CrossRef CAS PubMed;
(b) Z. D. Matović, V. D. Miletić, G. Samardzić, G. Pelosi, S. Ianelli and S. Trifunovi, Inorg. Chim. Acta, 2005, 358, 3135–3144 CrossRef PubMed.
|
| This journal is © The Royal Society of Chemistry 2015 |
Click here to see how this site uses Cookies. View our privacy policy here. 
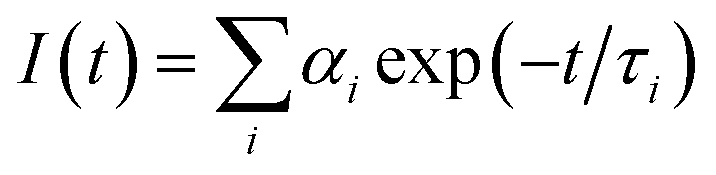 , where αi was a factor representing the fractional contribution to the time-resolved decay of the component with a lifetime of τi. Bi-exponential function was used to fit the decay profile for the complex, with obtaining χ2 close to 1. The intensity-averaged life time (τav) was determined from the result of the exponential model using
, where αi was a factor representing the fractional contribution to the time-resolved decay of the component with a lifetime of τi. Bi-exponential function was used to fit the decay profile for the complex, with obtaining χ2 close to 1. The intensity-averaged life time (τav) was determined from the result of the exponential model using  , where αi and τi are the pre-exponential factors and excited-state luminescence decay time associated with the i-th component, respectively.
, where αi and τi are the pre-exponential factors and excited-state luminescence decay time associated with the i-th component, respectively.