DOI:
10.1039/C4RA17306F
(Paper)
RSC Adv., 2015,
5, 16199-16206
Self-promoted phthalimide-containing phthalonitrile resins with sluggish curing process and excellent thermal stability†
Received
31st December 2014
, Accepted 23rd January 2015
First published on 23rd January 2015
Abstract
A novel unsymmetrical phthalimide-containing phthalonitrile (PIPN) was successfully synthesized. The chemical structure of the PIPN monomer was confirmed by various spectroscopic techniques. Rheology and differential scanning calorimetry (DSC) revealed that the self-promoted curing reaction of the PIPN was an extremely sluggish process. The fully cured PIPN polymers showed excellent thermal properties revealed by thermogravimetric analysis (TGA) and DSC. IR and solid-state UV-Vis diffusion reflectance spectra confirmed the formation of the triazine and phthalocyanine ring during the curing reaction. Rheometric studies suggested that the curing reaction of phthalonitrile with a phthalimide group was faster compared to the reaction with a benzimidazole ring, and a nucleophilic addition reaction mechanism was successfully introduced to explain this phenomenon.
1. Introduction
Phthalonitrile polymers were first introduced by Keller and co-workers as a class of high-temperature thermosetting polymers.1 Because of their outstanding thermal and thermal-oxidative stabilities, excellent mechanical and physiochemical properties, adjustable processability, and superior flame, water and chemical resistance, phthalonitrile polymers have been used for a wide range of applications in many fields, such as marine, aerospace and electronic packaging applications.1–6
The polymerization of the neat phthalonitrile resins is an extremely sluggish process which can be promoted by various curing agents4,5,7 such as aromatic diamines,4 aromatic phenols,8 and metallic salts.9 In previous works, much effort has been focused on the design of curing agents to improve the processability and performance of phthalonitrile resins. Typically, phthalonitrile resins are produced by heating phthalonitrile monomers in the presence of a certain percentage of curing additives for an extend period of time at elevated temperatures. These curing additives are always low molecular weight compounds and their volatility will reduce the mechanical properties of the products.4 To solve this problem, the amino and phenol groups were introduced into phthalonitrile monomers in our previous reports.10–12 The systematic investigation on the thermal polymerization behaviors of these amino and phenol-containing phthalonitrile monomers indicated a self-promoted polymerization behavior, and different curing behaviors of phenol-containing phthalonitrile monomers may be correlated to their different acidities.12 However, the introduction of amino or phenol groups to phthalonitrile resulted in undesirable thermal properties.11,12
According to our previous work, much work help us to conclude that the polymerization of phthalonitrile can be promoted by active hydrogen proton-containing groups, e.g. a benzimidazole ring (BI) with active hydrogen proton was introduced into phthalonitrile monomers (BIPN). The systematic investigation of BIPN indicated that introduction of benzimidazole is effective in maintaining the outstanding thermal properties of phthalonitrile resins and an extremely sluggish curing behavior is unexpectedly observed, which is favorable in fabricating thick composite components by resin infusion methods (RIM) or resin transfer molding (RTM).7 Inspired by the results, one can rationally imagine that introduction of active hydrogen-containing heterocyclic structures (AHCH) such as BI may be a common and effective method to design a kind of thermally stable and unique curing agents. On the other hand, the molecular mechanism on the catalytic activity of the AHCH is unclear. We also believe the effort to interpret this mechanism will both enrich the species of the curing agents of the phthalonitrile resins and promote the research on phthalonitrile curing mechanism.
As a part of an ongoing project dealing with the development of stable and efficient curing agents for phthalonitrile resins, a thermally stable phthalimide heterocyclic structure with active hydrogen was introduced into phthalonitrile monomer (phthalimide-containing phthalonitrile, PIPN). The curing behaviors, thermal behaviors and structure of PIPN were investigated systematically. The PIPN monomer showed faster self-promoted process compared to the corresponding BIPN model monomer, and a rational nucleophilic addition mechanism was introduced to explain this phenomenon. The explanation indicates that the basicities of the AHCH is an important factor in determining the polymerization rates of phthalonitrile resins. Additionally, this paper shows an efficient pathway to synthesis unsymmetrical compounds using symmetrical dianhydrides, e.g. 4,4′-oxydiphthalic anhydride (ODPA).
2. Experimental
Materials
4-Nitrophthalonitrile was purchased from Ji'nan Weido Chemical Co. Ltd. 3-Aminophenol was purchased from Sinophiarm Chemical Reagent Co. Ltd. ODPA was purchased from Shanghai Research Institute of Synthetic Resins. N,N′-Dimethylformamide (DMF), N-methyl-2-pyrrolidone (NMP), pyridine and potassium carbonate were purchased from Tianjin BoDi Co. Ltd. DMF and NMP were purified by distillation under reduced pressure over calcium hydride. Acetic anhydride was purchase from Chongqing Chuandong Chemical (Group) Co. Ltd. Urea and other chemicals were purchased from Chengdu Kelong Chemical Reagent Co. Ltd. and were all used as received.
Synthesis of PIPN
Synthesis of 4-(3-aminophenoxy) phthalonitrile. 4-(3-Aminophenoxy) phthalonitrile was synthesized according to a published literature.6
Synthesis of compound 1. To a 500 mL, three-necked flask was added ODPA (31.02 g, 0.10 mol) and dry NMP (124 mL). while stirring, the mixture was heated at 70 °C until the ODPA completely dissolved under an inert atmosphere, and then the solution of 4-(3-aminophenoxy) phthalonitrile (23.5 wt% in dry NMP, 23.52 g, 0.10 mol) was added dropwise (3–5 seconds per drop, as slowly as possible), after the solution was cooled to room temperature (20 °C). The reaction mixture was stirred for 6 h at room temperature after the solution of 4-(3-aminophenoxy) phthalonitrile was completely added, and then acetic anhydride (98.5 wt% 62.19 g, 0.60 mol) and pyridine (99.5 wt% 47.70 g, 0.60 mol) was added in the reaction solution. The reaction solution was heated to 120 °C and stirred for 3 h. Thin layer chromatography (TLC) was applied to monitor the synthetic reaction and determine the reaction extent since its completion is conveniently identified by the elimination of 4-(3-aminophenoxy) phthalonitrile.The reaction solution was then poured into methanol (2250 mL) and stirred for 1 hour and then filtered. The methanol in the filtrate was concentrated on a rotary evaporator at 45 °C. The leaving solution was washed with water, and the precipitate was collected by suction filtration. The solid was washed with water, filtered for 3 times, and then dried at 70 °C under reduced pressure for 12 h. Yield: 24.90 g 45.66%
Synthesis of compound 2. To a 100 mL, single-necked flask was added compound 1 (16.00 g, 0.029 mol) and acetic anhydride (98.5 wt% 13.00 g, 0.125 mol). The mixed solution was heated and refluxed for 1 h, and then cooled to room temperature. Then the acetic anhydride was concentrated on a rotary evaporator leaving a viscous solid.
Synthesis of PIPN
To a 100 mL, three-necked flask was added compound 2 (14.50 g, 0.027 mol) and dry DMF (6.46 g), and then urea (1.07 g, 0.018 mol) was added. The reaction solution was heated to 144–146 °C and stirred for 50 min. Then the reaction solution was washed with water after the reaction solution cooled to room temperature. The resulting precipitate (PIPN) was collected by filtration and purified by methanol reflux (2 h). Yield: 8.05 g, 57.84%. Then the powder of PIPN (5.00 g) was recrystallized from acetonitrile (200 mL). Yield: 3.52 g, 68.14%. IR (KBr, cm−1): 3225 (N–H stretching), 2234(C![[triple bond, length as m-dash]](https://www.rsc.org/images/entities/char_e002.gif) N stretching), 1770 (C
N stretching), 1770 (C![[double bond, length as m-dash]](https://www.rsc.org/images/entities/char_e001.gif) O stretching), 1720 (CO stretching), 1368 (C–N stretching). 1H NMR (300 MHz, DMSO-d6, δ): 11.43 (H, s, NH), 8.17–8.14 (H, d, Ar H), 8.06–8.03 (H, d, Ar H), 7.93 (H, s, Ar H), 7.91–7.90 (H, d, Ar H), 7.68–7.60 (3H, m, Ar H), 7.55–7.50 (2H, dd, Ar H), 7.48–7.28 (4H, m, Ar H). ELEM. ANAL: found: C, 67.93; N, 10.56; H, 2.55. Calcd for C30H14N4O6: C, 68.44; N, 10.64; H, 2.68%.
O stretching), 1720 (CO stretching), 1368 (C–N stretching). 1H NMR (300 MHz, DMSO-d6, δ): 11.43 (H, s, NH), 8.17–8.14 (H, d, Ar H), 8.06–8.03 (H, d, Ar H), 7.93 (H, s, Ar H), 7.91–7.90 (H, d, Ar H), 7.68–7.60 (3H, m, Ar H), 7.55–7.50 (2H, dd, Ar H), 7.48–7.28 (4H, m, Ar H). ELEM. ANAL: found: C, 67.93; N, 10.56; H, 2.55. Calcd for C30H14N4O6: C, 68.44; N, 10.64; H, 2.68%.
Preparation of PIPN polymer
The PIPN polymer was prepared by following heating process; 305 °C for 6 h, 325 °C for 2 h, 350 °C for 2 h and then post-cured at 375 °C for 2 h.
Characterization
Elemental analyses (EA) were performed on an Italy CARLO ERBA 1106 elemental analyzer. 1H NMR (300 MHz) was measured on a Bruker Avance-300 NMR spectrometer with DMSO-d6 as the solvent and tetramethylsilane as the internal standard. IR spectra were recorded with a Nicolet FTIR-380 Fourier transform infrared spectrometer by KBr pellet. The solid-state UV-Vis diffusion reflectance spectrum of the sample was analyzed with a UV-Vis spectrophotometer (UV-3600, Shimadzu, Japan). Thermal gravimetric analysis (TGA) and derivative thermogravimetric analysis (DTG) were carried out from 30 °C to 800 °C using a TA instrument Q500 thermogravimetric analyzer in flowing nitrogen of flowing air (60 mL min−1) at a heating rate of 10 °C min−1. Differential scanning calorimetry (DSC) was performed on a TA instrument Q200 differential scanning calorimeter under nitrogen atmosphere (50 mL min−1) at a heating rate of 10 °C min−1. Rheological experiments were conducted on a TA instruments AR-2000 rheometer in conjunction with an environmental testing chamber for temperature control. The measurements were made using a 25 mm diameter parallel plates at low strain values (2.5 × 10−4) and a frequency of 1 Hz. The sample specimen disks, 25 mm in diameter, were prepared by press-molding the BIPN powder (around 0.50 g) at room temperature. The compacted sample disk was subsequently loaded in the rheometer fixture. The complex viscosity as a function of temperature was measured from 30 to 360 °C at a heating rate of 5 °C min−1 in air. The complex viscosity of PIPN was also monitored at 295, 305, and 325 °C in air as a function of time. The morphology of the PIPN polymer was determined by a JEOL JSM-7500F field-emission scanning electron microscope (SEM). The specimen was coated with Au prior to observation.
Molecular simulation details for PIPN and BIPN monomer
The molecular simulation was applied to calculate the charge density on PIPN and its corresponding model compound BIPN. All calculations, including geometry optimizations for PIPN and BIPN structures were performed with the B3LYP exchange correlation corrected functional with the 6-31+g(d,p) basis set using the Gaussian 09W package.
Water uptake test
The PIPN polymer used for water uptake test was prepared by heating PIPN monomer at 305 °C for 6 h, 325 °C for 2 h, 350 °C for 2 h and then post-cured at 375 °C for 2 h in air condition. The sample of polymer was then immersed in boiling distilled water at 96 °C for 35 hours. The sample was removed from the boiling water, dried with filter paper, and weighed periodically to determine the amount of water absorbed.
3. Results and discussion
Monomer synthesis
The novel phthalimide-containing phthalonitrile, PIPN, was synthesized by a three-step reaction as illustrated in Scheme 1. Precise controlled condensation between ODPA and 4-(3-aminophenoxy) phthalonitrile using acetic anhydride and pyridine as dehydration and cyclization catalysts in NMP afforded compound 1. Dehydration and cyclization in compound 1 using acetic anhydride as dehydration and cyclization catalyst and solvent afforded compound 2. The PIPN was readily synthesized by reaction between compound 2 and urea using DMF as solvent. The chemical structure of the PIPN monomer was confirmed by IR, 1H NMR and EA.
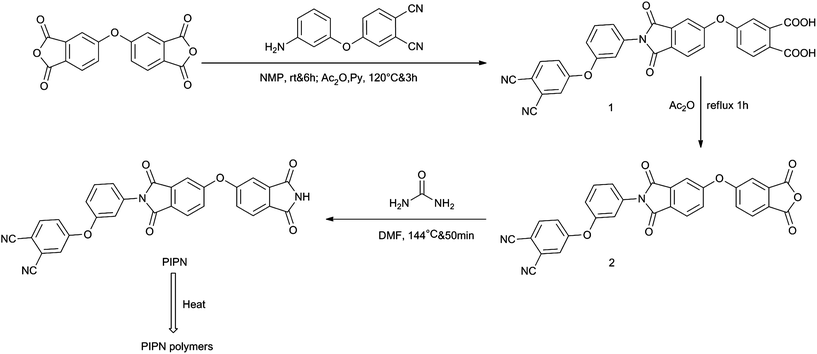 |
| | Scheme 1 Synthesis of PIPN monomer and polymers. | |
Polymerization behavior of PIPN
The thermal behaviors of the PIPN monomer and polymers were studied by DSC and TGA. The DSC first and second scan of PIPN monomer are shown in Fig. 1A and B. The first scan of DSC thermogram exhibited a smaller endotherm around 156 °C and a bigger endotherm around 210 °C, corresponding to loss of crystalline water and melting transition of PIPN monomer, respectively. The loss of crystalline water (smaller endotherm) can be confirmed by its TGA and EA (see ESI Fig. S2 and Table S1†). The second scan of DSC thermogram exhibited only a glass transition around 109 °C, and neither crystallization peak nor melting peak was observed. The absence of a crystallization or melting peak indicated that the PIPN proceeded somewhat self-propagating reaction during the heating run.13 Neither curve showed any thermal effects at elevated temperatures due to curing, which indicated that the polymerization of PIPN proceeded extremely sluggish and showed very low polymerization exothermic effect. Following the heat treatment (325 °C, 5 h) in the DSC (see ESI Fig. S3†), the sample also did not show any thermal effects, implying that the polymerization of PIPN was an extremely sluggish process without any obvious polymerization exothermic effect which can be used to fabricate thick composite components by RIM or RTM. The thermal behaviors of PIPN polymer were also studied by DSC (Fig. 1C). The PIPN polymer used for DSC measurements was cured at 305 °C for 6 h, 325 °C for 2 h, 350 °C for 2 h, 375 °C for 2 h in nitrogen did not exhibit any glass transition when the temperature was up to 400 °C, indicating that the post cured PIPN polymer exhibited excellent thermal properties.
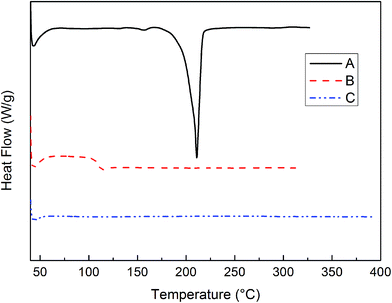 |
| | Fig. 1 DSC curves of PIPN under nitrogen atmosphere: (A) first scan of PIPN monomer (B) second scan of PIPN monomer (C) second scan of PIPN polymer. | |
The polymerization behaviors of PIPN were studied by rheological measurements. The viscosity of the PIPN was monitored as a function of temperature from 30 to 360 °C (see ESI Fig. S4†). The PIPN monomer showed a rapid decrease in viscosity on 210 °C, corresponding to the melting of PIPN monomer. The viscosity curve did not show any distinctive increase within the temperature range of 210–360 °C, which is consistent with the DSC results. Low melt viscosity and large processing window are useful for fabricating thick composite sections where the melt has to impregnate thick fiber preforms.
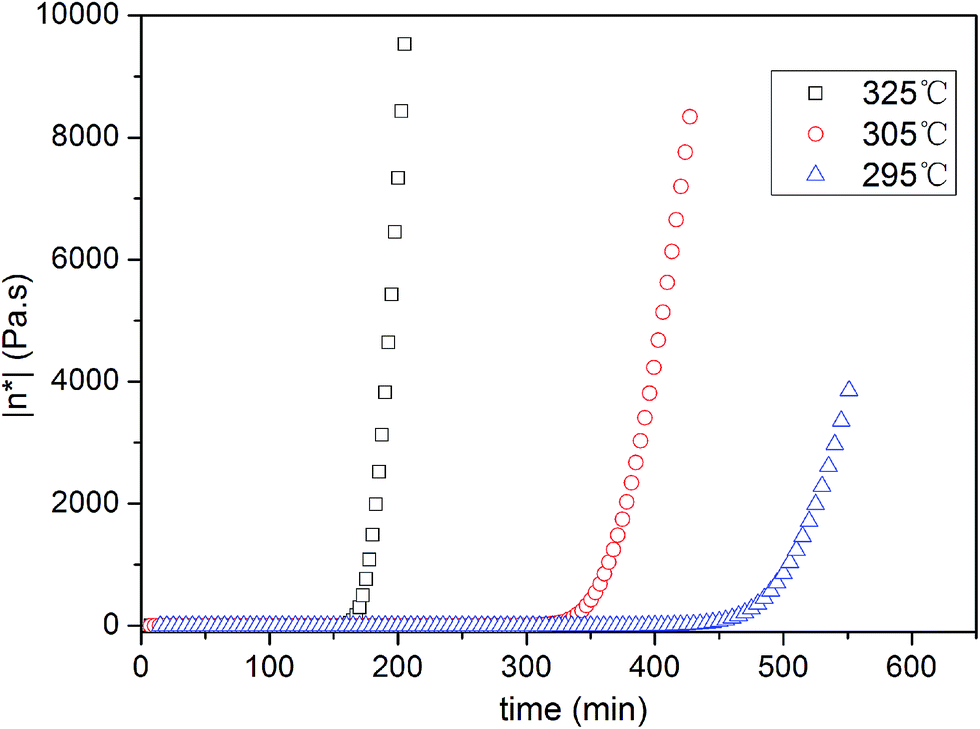
Rheometric studies on PIPN monomer were conducted to monitor the viscosity changes accompany the curing reaction. Viscosity increase as a function of reaction time for the polymerization of PIPN monomer at 295 °C, 305 °C and 325 °C are shown in Fig. 2 (as a comparison, see ESI Fig. S5†). From the plots it is evident that the low melt viscosities of PIPN can be kept for a very long time. The PIPN did not show a distinctive increase in viscosity within 150 min dwell at 325 °C, 300 min at 305 °C, and 450 min at 295 °C. On the other hand, the viscosities of PIPN increased rapidly after 160 min dwell at 325 °C, 350 min at 305 °C, and 500 min at 295 °C. Based on this study, the polymerization of PIPN is an extremely slower process comparing with the polymerization process of phthalonitrile which is promoted by aromatic amines or aromatic phenols,4,7,14 but it is a faster process comparing with the corresponding model compound BIPN (Fig. 3 and 4).7 It has been proposed that the initial curing reaction occurs via the protonation of cyano group nitrogen followed by nucleophilic attack by base at the carbon (Scheme 2).13,15–19 K. Zeng et al. stated that the acidities of hydroxyl-containing phthalonitrile may contribute to the different curing behaviours, supporting the proposed probably initial curing mechanism.12 The 1H NMR spectra of PIPN and BIPN are shown in Fig. 5, the chemical shift of active hydrogen in BIPN (12.98) is higher than PIPN (11.43), indicating a higher acidity of BIPN than that of PIPN. Therefore, referring to the AHCH promoted curing system, the higher acidity may lead to a higher curing rate. However, the fact is opposite. T. M. Keller et al. proposed that the different reactivities of primary amines toward phthalonitrile monomer may be correlated to the different basicities of primary amines.4,13,15 However, for the AHCH, due to their amphoteric nature, the relationship between structures and reactivities is still a puzzle. In this case, we proposed that the basicities of the AHCH moieties may be an important factor in determining the curing rates. To evaluate the basicities of the AHCH moieties in the PIPN and BIPN, the molecular simulation was conducted to calculate the charge density on their N atoms using Gaussian 09W package (Fig. S6 and S7†). The calculated absolute charge density of N atom of the PIPN (0.410) is higher than the BIPN (0.399), which indicates that the AHCH moiety in PIPN possesses stronger basicity than that of BIPN. Therefore, the AHCH moiety in PIPN possesses stronger nucleophilic ability than in BIPN. The consistence of molecular simulation and experiment further confirmed the existence of a nucleophilic addition process during the polymerization of phthalonitrile resins. Therefore, the different reactivities of AHCH toward phthalonitrile may be also correlate to their different basicities. Based on this study, a rational molecular design scheme of efficient curing agents for phthalonitrile resins can be developed.
 |
| | Fig. 2 Complex viscosity as a function of time for the PIPN at various temperatures. | |
 |
| | Fig. 3 The structure of BIPN. | |
 |
| | Fig. 4 Complex viscosity vs. time curve at 325 °C for (□) PIPN and (▽) BIPN. | |
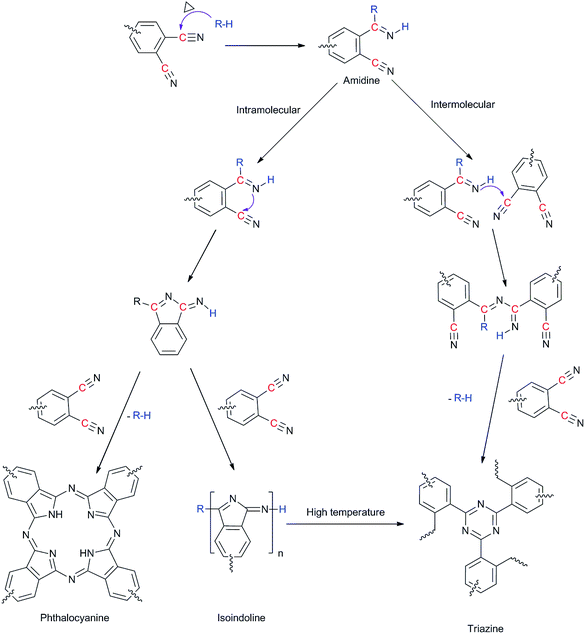 |
| | Scheme 2 Widely accepted curing mechanism of phthalonitrile resins. | |
 |
| | Fig. 5 1H NMR of PIPN and BIPN. | |
Characterization of polymers
The PIPN monomer and polymers were characterized by FTIR spectra (Fig. 6) and solid-state UV-Vis spectra (Fig. 7). The PIPN polymers, which were used for FTIR spectra and solid-state UV-Vis spectra, were obtained by prolonged heating at 325 °C in high-temperature oven under air atmosphere. The nitrile characteristic absorption band was observed at 2234 cm−1 in the FTIR spectra of the PIPN monomer and polymer. The nitrile absorption of the PIPN polymer, which was about 20% of the origin intensity of the PIPN monomer, decreased with the polymerization at 325 °C for 10 h (Fig. 6), normalized to the lactam carbonyl bands at 1720 cm−1 and 1770 cm−1. Instead, the intensity of the absorption of CN stretch around 1527 cm−1 increased obviously, indicating the formation of triazine ring during the polymerization process. The emergence of extremely small peak around 1010 cm−1 was possibly contributed to the formation of phthalocyanine ring.20,21 The phthalocyanine ring was also clearly identified by UV-Vis reflection at 700 nm as shown in a representative solid-state UV-Vis spectra of PIPN polymers (Fig. 7, and ESI S8†).21 Both the relative absorptions of the phthalocyanine ring in FTIR and solid-state UV-Vis spectra were extremely weak, indicating that the formation of phthalocyanine during the polymerization of PIPN was a very weak process.22 These results are consistent with the proposed polymerization process which is illustrated in Scheme 2.
 |
| | Fig. 6 FTIR spectra of PIPN (a) monomer (b) polymer. | |
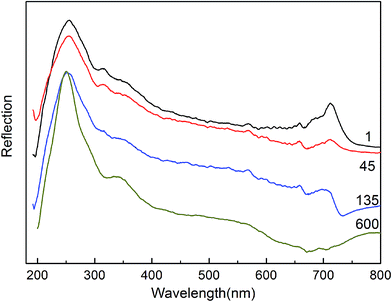 |
| | Fig. 7 Solid-state UV-Vis reflectance spectra of PIPN cured at 325 °C for a prolonged time (1) 1 min (45) 45 min (135) 135 min (600) 600 min. | |
Thermal properties of PIPN
The PIPN polymers were obtained by heating at 305 °C for 6 h, 325 °C for 2 h, 350 °C for 2 h, 375 °C for 2 h in TGA chamber under nitrogen atmosphere with a flow rate of 60 mL min−1. The thermal properties of PIPN monomer and polymers were evaluated by TGA (Fig. 8) and DTG (Fig. 9) in air and nitrogen atmosphere, and the specific parameters were collected in Table 1. The PIPN polymer exhibited excellent thermal property with 68.6% weight retention approximately at 800 °C in nitrogen atmosphere, and it showed the weight residual of 99% (T1%), 95% (T5%), 90% (T10%) at 476.3 °C, 517.7 °C and 542.6 °C respectively. It also exhibited outstanding stability in air atmosphere as shown in the TGA curves (Fig. 8) and Table 1, and it showed the T1%, T5% and T10% at 481.0 °C, 518.6 °C and 539.6 °C respectively. The results of these studies indicated that the PIPN polymers exhibited excellent thermal and thermo-oxidative stabilities.11,12 The corresponding DTG curves of PIPN monomer and polymers in nitrogen and air atmosphere are displayed in Fig. 9, the maximum decomposition peak of PIPN polymer in air might be ascribed to the decomposition of ether linkages and phenyl structures, the evaporation of phthalocyanine and triazine and phthalocyanine-containing small molecules.16 The decomposition peaks of PIPN monomer at 436 °C in nitrogen and air might be ascribe to the evaporation of the PIPN monomer, because the decomposition temperatures of PIPN polymers is much higher than 436 °C (Fig. 8 and 9). Unexpectedly, the DTG peak at 436 °C of PIPN monomer in air is smaller than its corresponding peak in nitrogen. This unique phenomenon cannot be ascribed to the oxidation of PIPN monomers, since the corresponding model compounds of PIPN, phthalonitrile terminated ODPA monomer (CNODPA, see ESI Fig. S1†) and phthalimide terminated ODPA monomer (HIODPA, see ESI Scheme S1†), neither show this unique phenomenon before 450 °C (see ESI Fig. S9 and S10†), and the TGA and DTA curves of PIPN polymers also (Fig. 8 and 9) confirmed the outstanding thermo-oxidative stability of the groups in PIPN monomer. This unique phenomenon might be induced by relatively faster polymerization rate in air condition which inhibited the volatile of small molecules. Kishore et al. stated that oxygen could accelerate the radical polymerization under certain conditions such as high temperatures.23,24 Inspired by these statements, it is rational to speculate that the radical reaction might played an important role at high temperatures in the polymerization of phthalonitrile resins such as the PIPN resin (see ESI Fig. S10 and S11†). This unique phenomenon will be further discussed in our following research.
 |
| | Fig. 8 TGA curves of PIPN monomer and polymers. | |
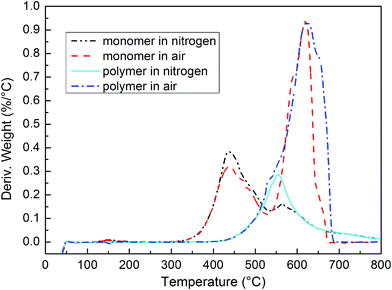 |
| | Fig. 9 DTG curves of PIPN monomer and polymers. | |
Table 1 The TGA data of PIPN monomer and polymers T1%, T5% and T10% mean weight loss were 1%, 5% and 10%, respectively
| BIPN |
T1% (°C) |
T5% (°C) |
T10% (°C) |
Char yield (%) |
| Monomer in nitrogen |
366.9 |
405.9 |
425.1 |
43.2 |
| Monomer in air |
372.9 |
407.8 |
427.2 |
3.1 |
| Polymer in nitrogen |
476.3 |
517.7 |
542.6 |
68.6 |
| Polymer in air |
481.0 |
518.6 |
539.6 |
2.6 |
In addition, the surface morphology of the PIPN polymer are shown in Fig. 10. No voids can be observed for the PIPN polymer at different magnifications, which confirms the void-free structure of PIPN polymer and it also guarantees the excellent thermal and mechanical properties. Moreover, the addition polymerization mechanism and low volatility of the PIPN monomer are two most important factors for preparing void-free PIPN polymer.
 |
| | Fig. 10 SEM images of PIPN polymer surfaces. (a) ×1000, (b) ×5000. | |
Water uptake
The water uptake was calculated using the following equation:
| Water uptake (%) = (M2 − M1)/M1 × 100 |
where, M1 and M2 are the weight of the PIPN polymer at dry state and after immersion in boiling distilled water.
Fig. 11 shows the water uptake properties of PIPN polymer in boiling distilled water. As shown in Fig. 11, the water uptake increases rapidly during the first stage, and it appears to level off after approximately 27 hours in boiling distilled water. Its water uptake is comparable to other phthalonitrile-based polymers.16,25,26 It is well recognized that degree of crosslinking and contents of hydrophilic groups on molecular chains are two important factors influencing water uptake properties of a polymer16,26 For the PIPN polymer, the comparable water uptake can be attributed to its high crosslinking degree and void-free structure, although it contains hydrophilic imide moiety. Furthermore, the water uptake performance of phthalimide-promoted phthalonitrile can be enhanced by controlling the content of hydrophilic imide groups in a rational designed phthalonitrile systems.
 |
| | Fig. 11 Water uptake of PIPN polymer in boiling distilled water. | |
4. Conclusions
A novel unsymmetrical phthalimide-containing phthalonitrile has been successfully synthesized. Phthalimide heterocyclic structure with active hydrogen proton is responsible for the self-promoted polymerization reactions of the PIPN. The cured PIPN polymers showed outstanding thermal and thermo-oxidative stabilities. The sluggish curing process and low polymerization exothermic of PIPN are favorable in fabricating thick composite components by RIM or RTM. The PIPN monomer showed faster self-promoted process compared to the corresponding BIPN model monomer. This might be attributed to the differences in the relative nucleophilic reactivities of their AHCH moieties which are correlated to their different basicities. This will contribute to develop stable and efficient curing agents for phthalonitrile resins.
Acknowledgements
We thank the National Natural Science Foundation of China (no. 51173114) for the financial support.
Notes and references
- T. M. Keller and J. R. Griffith, ACS Symp. Ser., 1980, 25–34 CrossRef CAS PubMed.
- K. Zeng and G. Yang, Phthalonitrile Matrix Resins and Composites, Wiley Online Library, 2012 Search PubMed.
- A. P. Mouritz, E. Gellert, P. Burchill and K. Challis, Compos. Struct., 2001, 53, 21–42 CrossRef.
- S. B. Sastri and T. M. Keller, J. Polym. Sci., Part A: Polym. Chem., 1998, 36, 1885–1890 CrossRef CAS.
- D. D. Dominguez, H. N. Jones and T. M. Keller, Polym. Compos., 2004, 25, 554–561 CrossRef CAS.
- T. M. Keller and D. D. Dominguez, Polymer, 2005, 46, 4614–4618 CrossRef CAS PubMed.
- D. Wu, Y. Zhao, K. Zeng and G. Yang, J. Polym. Sci., Part A: Polym. Chem., 2012, 50, 4977–4982 CrossRef CAS.
- D. Augustine, D. Mathew and C. P. Nair, Polym. Int., 2013, 62, 1068–1076 CAS.
- D. R. Boston and J. C. Bailar Jr, Inorg. Chem., 1972, 11, 1578–1583 CrossRef CAS.
- K. Zeng, K. Zhou, W. R. Tang, Y. Tang, H. F. Zhou, T. Liu, Y. P. Wang, H. B. Zhou and G. Yang, Chin. Chem. Lett., 2007, 18, 523–526 CrossRef CAS PubMed.
- S. Zhou, H. Hong, K. Zeng, P. Miao, H. Zhou, Y. Wang, T. Liu, C. Zhao, G. Xu and G. Yang, Polym. Bull., 2009, 62, 581–591 CrossRef CAS PubMed.
- K. Zeng, K. Zhou, S. Zhou, H. Hong, H. Zhou, Y. Wang, P. Miao and G. Yang, Eur. Polym. J., 2009, 45, 1328–1335 CrossRef CAS PubMed.
- G. Yu, C. Liu, X. Li, J. Wang, X. Jian and C. Pan, Polym. Chem., 2012, 3, 1024–1032 RSC.
- A. Badshah, M. R. Kessler, Z. Heng, J. H. Zaidi, S. Hameed and A. Hasan, Polym. Chem., 2013, 4, 3617–3622 RSC.
- P. J. Burchill, J. Polym. Sci., Part A: Polym. Chem., 1994, 32, 1–8 CrossRef CAS.
- F. Zhao, R. Liu, C. Kang, X. Yu, K. Naito, X. Qu and Q. Zhang, RSC Adv., 2014, 4, 8383–8390 RSC.
- M. J. Sumner, R. Y. Weyers, A. C. Rosario, J. S. Riffle and U. Sorathia, Polymer, 2004, 45, 5199–5206 CrossRef CAS PubMed.
- M. J. Sumner, M. Sankarapandian, J. E. McGrath, J. S. Riffle and U. Sorathia, Polymer, 2002, 43, 5069–5076 CrossRef CAS.
- D. Augustine, K. P. Vijayalakshmi, R. Sadhana, D. Mathew and C. P. Reghunadhan Nair, Polymer, 2014, 55, 6006–6016 CrossRef CAS PubMed.
- M. Laskoski, A. Neal, T. M. Keller, D. Dominguez, C. A. Klug and A. P. Saab, J. Polym. Sci., Part A: Polym. Chem., 2014, 52, 1662–1668 CrossRef CAS.
- L. Wang, J. Li, W. Zhang, G. Chen, W. Zhang and X. Zhu, Polym. Chem., 2014, 5, 2872–2879 RSC.
- T. M. Keller, Chem. Mater., 1994, 6, 302–305 CrossRef CAS.
- V. A. Bhanu and K. Kishore, Chem Rev., 1991, 91, 99–117 CrossRef CAS.
- A. K. Nanda, J. Jayaseharan and K. Kishore, J. Appl. Polym. Sci., 2001, 79, 1549–1555 CrossRef CAS.
- S. B. Sastri and T. M. Keller, J. Polym. Sci., Part A: Polym. Chem., 1999, 37, 2105–2111 CrossRef CAS.
- H. Sheng, X. Peng, H. Guo, X. Yu, C. Tang, X. Qu and Q. Zhang, Mater. Chem. Phys., 2013, 142, 740 CrossRef CAS PubMed.
Footnote |
| † Electronic supplementary information (ESI) available. See DOI: 10.1039/c4ra17306f |
|
| This journal is © The Royal Society of Chemistry 2015 |
Click here to see how this site uses Cookies. View our privacy policy here.