
A lithium-ion oxygen battery using a polyethylene glyme electrolyte mixed with an ionic liquid†
Abstract
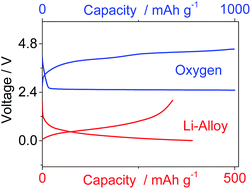
In this paper, we propose a new electrolyte formed by dissolving lithium-bis-(trifluoromethanesulfonyl)-imide (LiTFSI) salt in a long chain glyme, i.e. poly(ethylene glycol)500-dimethylether (PEG500DME) mixed with methyl butyl pyrrolidinium-bis-(trifluoromethanesulfonyl)-imide (Pyr14TFSI) ionic liquid. The electrolyte composition ensures both very low volatility and good performance as demonstrated by thermal analysis and electrochemical tests. The selected solution is efficiently employed as the electrolyte both in a lithium–oxygen half-cell, and in a full lithium-ion oxygen battery using a Li–Sn–C nanostructured composite anode, thus suggesting the suitability of the polyethylene glyme mixed with the ionic liquid for application in high-energy storage systems.
- This article is part of the themed collection: Ionic Liquids: Editors collection for RSC Advances
 Please wait while we load your content...
Please wait while we load your content...

