
Heavier chalcogenone complexes of bismuth(iii)trihalides: potential catalysts for acylative cleavage of cyclic ethers†
Abstract
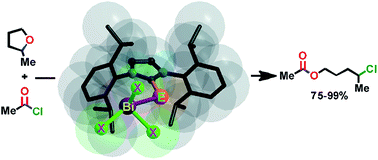
Heavier chalcogenones (S, Se and Te) of imidazole act as versatile ligands to yield a series of mononuclear and dinuclear bismuth(III)complexes of heavier chalcogenones in excellent yield. These new bismuth heavier chalcogen derivatives are the first structurally characterized molecules, where the bismuth and heavier chalcogen ratio is 1 : 1. There is only one previous report of a crystal structure of a bismuth(III)–imidazol selone compound and none with bismuth(III)–imidazol tellone. The bismuth center in monomeric bismuth chalcogen trihalides depicts pseudo trigonal bipyramidal geometry, while the dimeric bismuth chalcogen trihalides demonstrate distorted square pyramidal geometry. The solid state structures of bismuth chalcogenone derivatives feature rare Bi⋯π(aryl) interactions. Thus, the centroid of the C6-ring suggests a half sandwich type of bismuth environment in mononuclear and dinuclear bismuth(III) chalcogenone complexes. Notably, the Bi⋯π(aryl) interaction is not often noticed for mononuclear bismuth chalcogen compounds. Some of the bismuth(III) chalcogenone complexes also exhibit C–H⋯π(aryl), C–H⋯S and C–H⋯Cl types of hydrogen bonding. The bismuth–chalcogen bond distance in mononuclear bismuth(III)tribromide chalcogenone complexes is slightly longer than in mononuclear bismuth(III)trichloride chalcogenone complexes. A gradual increase in carbon–chalcogen bond distance was observed from the free imidazole–chalcogenone to mononuclear bismuth(III)trichloride chalcogenones, dinuclear bismuth(III)trichloride chalcogenones and mononuclear bismuth(III)tribromide chalcogenones and dinuclear bismuth(III)tribromide chalcogenones. The UV-vis absorption properties and thermal decomposition properties of imidazol chalcogenones and their bismuth derivatives were investigated. Furthermore, the O-acylative cleavage of cyclic ethers was demonstrated using mononuclear and dinuclear bismuth(III)complexes of heavier chalcogenones as catalysts. In contrast to bismuth(III)trichloride and bismuth(III)tribromide catalysts, mononuclear and dinuclear bismuth(III)complexes of heavier chalcogenones are very active towards an acylative cleavage of cyclic ethers through a mild and regioselective strategy. In particular, mononuclear imidazolthione–bismuth(III)trichloride is very active towards O-acylative cleavage of 2-methyl tetrahydrofuran.
 Please wait while we load your content...
Please wait while we load your content...

