
Intracellular pH-responsive mesoporous hydroxyapatite nanoparticles for targeted release of anticancer drug
Dalong Lia,
Jinmei Hea,
Xin Huanga,
Jiwei Lia,
Huayu Tianb,
Xuesi Chenb and
Yudong Huang*ac
aSchool of Chemical Engineering and Technology, Harbin Institute of Technology, Harbin, 150001, China. E-mail: 12B925006@hit.edu.cn; Fax: +86-451-86414806; Tel: +86-451-86414806
bKey Laboratory of Polymer Ecomaterials, Changchun Institute of Applied Chemistry, Chinese Academy of Sciences, Changchun, 130022, China
cState Key Laboratory of Urban Water Resource and Environment, Harbin Institute of Technology, Harbin, 150001, China
First published on 26th March 2015
Abstract
The design and synthesis of multifunctional nanocarriers is becoming a more and more interesting topic, and shows promising potential for clinical applications. Mesoporous hydroxyapatite nanoparticles (MHAPNs) have emerged as one of the most promising drug delivery vehicles due to their excellent performance. Herein, we constructed a novel cell-targeting, pH-sensitive nanocarrier based on MHAPNs for intracellular drug delivery (LA-BSA-CBA-MHAPNs), using lactobionic acid-conjugated bovine serum albumin (LA-BSA) molecules as end-caps, and 4-carboxyphenylboronic (CBA) as intermediate linkers. The constructed LA-BSA-CBA-MHAPNs as drug delivery carriers exhibited good loading capacity and high pH-controlled release efficiency. Under normal physiological conditions (pH 7.4), they showed a slow drug release rate, while under acidic subcellular environments (pH 5.0) the release rate was enhanced dramatically due to the breakage of cyclic ester bonds linkages between LA-BSA and MHAPNs which made the pores open. Furthermore, our study also demonstrated that the incorporation of the targeted group lactose acid played a positive role in enhancing HepG2 cell uptake efficiency in the LA-BSA-CBA-MHAPNs system in comparison to that of MHAPNs. All these results imply that the LA-BSA-CBA-MHAPNs system could play a significant role in constructing pH-responsive, controlled drug delivery systems for clinical therapy.
1 Introduction
In recent years, the construction of stimuli-responsive controlled-release systems for targeted drug delivery has attracted the interest of a broad range of researchers and provided a variety of promising applications in biomedical fields.1–3 Targeted drug delivery systems aim to improve the efficacy and reduce the toxicity of potent therapeutics in treatments for a variety of diseases. The release of guest molecules can be achieved in response to external stimuli such as temperature,4,5 pH,6–11 light,12–14 redox reagents,15,16 ultrasound,17,18 enzymes,19–21 etc. Among these, the pH-sensitive system is of special interest for cancer therapy given that both extracellular tumor (pH 6.5) and endosomes (pH 5.0) of cancer cells are more acidic than normal tissues (pH 7.4), which enables the carriers to release anticancer drugs in a pH dependent manner.22,23 Because of the acidic tumor microenvironment in solid tumors, the pH targeting approach is regarded as a more general strategy than many of other targeting approaches.24 Recently, many methods have been developed to synthesize pH-responsive drug delivery system to improve efficiency and reduce systemic toxicity of anticancer drugs.7–9 Among them, ordered mesoporous materials have gained much attention due to their unique surface and textural properties, and easy surface modifications for targeted delivery.25–27 However, until now, most of the reported models are designed based on the mesoporous silica nanoparticles as templates28–32 differing from this using other mesoporous nanoparticles as templates such as mesoporous hydroxyapatite particles to design the models should be an interesting topic.Hydroxyapatite [Ca10(PO4)6(OH)2] (HAP), which is the major component of bone and teeth, is a key biomaterial in view of its excellent biocompatibility, bioactivity, nontoxicity and noninflammatory.33,34 Mesoporous hydroxyapatite nanoparticles (MHAPNs) prepared by surfactant templating method35,36 posses attractive features such as large pore volumes, large surface area and reactive surfaces for post-functionalization which make them ideal as potential carriers for drugs. Moreover, it has also been confirmed that MHAPNs with the size less than 100 nm could be uptaken by cells efficiently. To explore such MHAPNs into a stimuli-responsive drug delivery models, the key point is how to seal the mesopores and “open” the sealants in response to external stimuli. Considering the acidic gradient environment in the tumor, pH-sensitive controlled-release system is of tremendous interest. At present, a number of controlled-release systems based MHAPNs with various “gatekeepers” have been investigated due to their biocompatibility, solubility and less toxicity than silica, quantum dot, and carbon nanotubes.37–41 Despite of these reports, the construction of an intelligent intracellular drug controlled-release system based on MHAPNs, in particular, considering the targeted release of the encapsulated anticancer drugs when passing through a region of low pH while keeping stable under physiological conditions to reduce side effects, remains a big challenge.
Herein, we prepared a novel pH-responsive controlled-release nanocarrier based on MHAPNs employing lactobionic acid-grafted bovine serum albumin (LA-BSA) molecules as efficient end-caps. After the drug loading, LA-BSA molecules were conjugated onto the surface of MHAPNs to close the pores with carboxyphenylboronic acid (CBA) molecules via an esterification reaction. The LA molecules act both as intermediate agents for conjugating with CBA via an esterification reaction and as ligands to confer the system target ability through the special interaction with galactose receptors of HepG2 cells.42 When cycle ester bonds which act as linkages between LA-BSA and MHAPNs are cleaved in acidic environment, the leave of LA-BSA makes the “gate” open and the encapsulated drug release.43–45 Considering these virtues, it is no doubt that this system shows great potential for both cell-specific targeting and intracellular pH-responsive release drug. To the best of our knowledge, this is the first report on biocompatible MHAPNs as carrier with LA-BSA gatekeepers covalently connected onto the particle via cycle ester bonds to construct intracellular pH-responsive controlled-release systems for targeted drug delivery.
2 Materials and methods
2.1 Materials
PEO99PPO65PEO99 (F127), N-hydroxysuccinimide (NHS), fluorescein isothiocyanate (FITC), 1-[3-(dimethylamino)propyl]-3-ethylcarbodiimide hydrochloride (EDC), doxorubicin hydrochloride (DOX·HCl), lactose acid (LA) and bovine serum albumin (BSA) were purchased from Sigma Chemical Co. (St. Louis, MO, USA). Calcium pantothenate (C18H32CaN2O10), dipotassium hydrogenphosphate trihydrate (K2HPO4·3H2O), 1,3,5-trimethylbenzene (TMB), 3-aminopropyltriethoxysilane (ARTS), 4-carboxyphenylboronic acid (CBA), trypan blue and triton X-100 were obtained from J&K Scientific Co. (Beijing, PR China). Human hepatocellular liver carcinoma cells (HepG2 cells), human umbilical vein endothelial cells (HUVECs), bovine serum, penicillin and streptomycin were provided by Sanggon biotech Co. (Shanghai, PR China). All the initial chemicals in the work were used without further purification.2.2 Preparation of MHAPNs
The MHAPNs were synthesized using the templating method based on a modified method in the literature.35 Block co-polymer pluronic F127 was selected as template, and TMB was used as pore-expanding agent. Firstly, 2.0 g of F127 and 10.12 g of calcium pantothenate (C18H32CaN2O10) were co-dissolved into 80 mL of distilled water and stirred vigorously 2 h, then 15 mL 1,3,5-trimethylbenzene (TMB) was added to form solution 1. In 40 mL of distilled water, 2.8 g of dipotassium hydrogenphosphate trihydrate (K2HPO4·3H2O) was dissolved to form solution 2. The pH of solution 2 was adjusted to 10 with ammonia (NH3·H2O). Finally, solution 2 was added dropwise to solution 1. The mixture solution was heated to 90 °C under reflux for 24 h and filtered to get precipitate. The white precipitate was subsequently dried in an oven at 100 °C for 48 h, then calcined at 600 °C for 6 h in muffle furnace.2.3 Preparation of CBA functionalized MHAPNs (CBA-MHAPNs)
Firstly, 2.1 g of MHAPNs and 8.5 mL of 3-aminopropyltriethoxysilane (ARTS) were dissolved in 120 mL toluene under stirring condition. The mixture solution was heated to 60 °C under reflux for 36 h and filtered to get precipitate. The white precipitate was subsequently dried at 100 °C for 48 h in a vacuum drying to get NH2-MHAPNs. Then, 0.4 g of CBA, 0.1 g of NHS and 0.2 g of EDC were co-dissolved in 70 mL of dimethyl sulfoxide (DMSO) and stirred vigorously 2 h. Subsequently, 1.25 g of NH2-MHAPNs was added to the solution and mixture solution was continuously stirred for 48 h under 25 °C. The solution was centrifuged after the reaction completed. The obtained precipitate was eluted with ethanol and water ten times to remove solvent. Finally the product was dried in a vacuum oven at 100 °C for 24 h to get CBA-MHAPNs.2.4 Preparation of LA functionalized BSA (LA-BSA)
1.0 g of LA, 0.15 g of NHS and 0.25 g of EDC were co-dissolved in 80 mL pH 5.0 PBS solution and stirred for 3 h. Subsequently, 1.2 g of BSA was added to above solution, and mixture solution was continuously stirred for 48 h. Finally the solution was vacuum freeze-dried for 24 h to get LA functionalized BSA (LA-BSA). LA was conjugated to the surface BSA molecules via an esterification reaction.2.5 The construction of LA-BSA end-capped pH-responsive MHAPNs systems (LA-BSA-CBA-MHAPNs)
DOX or FITC was utilized as both model drug and site marker for intracellular tracing of MHAPNs. Firstly, 6 mg of DOX or FITC and 0.15 g of CBA-MHAPNs were dissolved in 100 mL of distilled water and stirred for 48 h. Then 0.3 g of LA-BSA was added to the above mixture solution and stirred continuously for 36 h. By esterification reaction of hydroxy groups of LA and boronic acid groups of CBA-MHAPNs, pH-responsive MHAPNs nanoreservoirs end-capped with LA-BSA was constructed. The mixture solution was centrifuged and eluted with ethanol and water ten times to remove unencapsulated DOX or FITC. Cell-specific targeting moiety LA-BSA-CBA-MHAPNs was constructed after vacuum freeze-dried for 36 h.2.6 Drug loading and release assay
The LA-BSA-CBA-MHAPNs was used as carrier for DOX. End-capping efficiency and release behavior of LA-BSA-CBA-MHAPNs@DOX were detected by UV-vis fluorescence spectroscopy. 3.3 mg of LA-BSA-CBA-MHAPNs@DOX powders was dispersed into 10 mL of phosphate buffer solution (PBS) at pH 7.4, 6.5 or 5.0, respectively. The dispersion was transferred into a dialysis bag (molecular weight cut-off 8000 g mol−1), and then the bag was immersed into 100 mL of PBS solution with the same pH conditions. The volume of the dissolution media was maintained at 100 mL at 37 °C. 1.0 mL of solution was withdrawn at a given time interval, followed by supplying the same volume of fresh PBS solution. The amount of released drug in the PBS solution was measured by UV-vis spectrophotometer at 480 nm. In order to investigate the release of rapid responsive, another assay, 3.3 mg of LA-BSA-CBA-MHAPNs@DOX was suspended in 10 mL of PBS (pH 7.4). The suspension was transferred into a dialysis bag (molecular weight cut-off 8000 g mol−1), and subsequently placed in a beaker containing 100 mL of PBS buffer with the same pH conditions for 5 h. Then the pH of solution was adjusted to pH 5.0. At a predetermined time, 1 mL of nanoparticle suspension was withdrawn, followed by supplying the same volume of fresh PBS solution. The supernatant was taken for UV-vis spectrophotometer at 480 nm to determine the amount of released DOX.2.7 Cell culture
HepG2 cells and HUVECs were cultured in low-glucose Dulbecco's Modified Eagle's Medium supplemented with 10% bovine serum (FBS; Gibco), 100 U mL−1 of penicillin and 100 μg mL−1 of streptomycin in a 5% CO2 incubator at 37 °C under 95% humidity, respectively. Cell culture media was changed every 2 days. When reaching confluence, cells were detached with 0.25% trypsin in 1 mM of tetrasodium EDTA, centrifuged and resuspended in complete medium for reseeding in new culture flasks.2.8 Cytotoxicity assays
Cell viability was determined by the MTT assays. HepG2 cells seeded on 24-well plates with an initial seeding density of 2 × 104 cells cm2. When the cells reached 60–70% confluence, 20 μg mL−1 of MHAPNs and LA-BSA-CBA-MHAPNs were added to co-culture, respectively. After culture 6 h, 12 h and 24 h, cells were rinsed with PBS solution and changed with fresh culture medium. About 0.1 mL of MTT (5 mg mL−1) was added to each well and incubated in the CO2 incubator at 37 °C for another 4 h. Then, MTT containing medium was removed and 0.5 mL of dimethyl sulfoxide (DMSO) was added into each well to dissolve the formazan crystals that had formed. Absorbance values of formazan were determined with Bio-Rad model-680 microplate reader at 490 nm. Six replicates were done for each treatment group.2.9 Confocal laser scanning microscopy (CLSM) observation
FITC exhibits intense green fluorescence under UV light. This property allows the use of fluorescence to study the distribution of LA-BSA-CBA-MHAPNs inside the HepG2 cells. A suspension 20 μg mL−1 of LA-BSA-CBA-MHAPNs in PBS was introduced to culture medium overnight to mimic the blood circulation process prior to the cellular uptake. After incubation with nanoparticles in medium for 6 h, 12 h and 24 h, cells were washed for five times with pH 7.4 PBS to remove the residual nanoparticles. Then 200 μg mL−1 of trypan blue was added to quench fluorescence of extracellular for 10 min, respectively. Cells were fixed with 2% glutaraldehyde at 4 °C for 20 min, the fixed samples were then rinsed with excessive PBS buffer and permeabilized with 0.2% Triton X-100 at 4 °C for 10 min. The nuclei of cells were stained with 10 μg mL−1 of Hoechst 33258 at 4 °C for 10 min. Finally, the stained samples were mounted with 90% glycerine. The distribution of fluorescently labeled nanoparticles in HepG2 cells was observed with confocal laser scanning microscopy (CLSM).2.10 Flow cytometry
The number of HepG2 cells and endothelial cells, which engulfed MHAPNs and LA-BSA-CBA-MHAPNs, was detected by flow cytometry. Flow cytometry analysis was performed on a FACS (Becton Dickinson Immunocytometry System, San Jose, CA) by counting 10![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) 000 events. HepG2 cells and HUVECs were cultured on 24-well plates at an initial density of 2 × 104 cells cm2, respectively. After 7 days, 20 μg mL−1 of MHAPNs@FITC and LA-BSA-CBA-MHAPNs@FITC with the equivalent FITC concentrations were added to co-incubate and treated by the same procedures mentioned in CLSM observation section. After culture 6 h, 12 h and 24 h, cells were then rinsed with excessive PBS buffer to remove the unbound nanoparticles. Then 200 μg mL−1 of trypan blue was added to quench fluorescence of extracellular for 10 min, respectively. The collected cells were then dispersed in 1 mL of fresh PBS buffer for flow cytometry analysis with BD FACS Aria.
000 events. HepG2 cells and HUVECs were cultured on 24-well plates at an initial density of 2 × 104 cells cm2, respectively. After 7 days, 20 μg mL−1 of MHAPNs@FITC and LA-BSA-CBA-MHAPNs@FITC with the equivalent FITC concentrations were added to co-incubate and treated by the same procedures mentioned in CLSM observation section. After culture 6 h, 12 h and 24 h, cells were then rinsed with excessive PBS buffer to remove the unbound nanoparticles. Then 200 μg mL−1 of trypan blue was added to quench fluorescence of extracellular for 10 min, respectively. The collected cells were then dispersed in 1 mL of fresh PBS buffer for flow cytometry analysis with BD FACS Aria.
2.11 Characterization
A series of characterization technique was used to analyze the structural properties of each product. The morphology and mesoporous aperture of nanoparticles were observed by transmission electron microscopy (TEM; Philips EM20). The structure of nanoparticles was analyzed by Fourier transform infrared (FTIR; VECTOR22, BRUKER) spectrum within the scanning range of 4000–400 cm−1 using the KBr pellet technique. Powder X-ray patterns (XRD) were recorded on a Bruker D4 X-ray diffractometer with Ni-filtered Cu Kα radiation (40 KV, 40 mA). N2 adsorption–desorption isotherms were measured with an automatic surface area and porosity analyzer (3H-2000PS2, Beishide) at 77 K. The Brunauer–Emmett–Teller (BET) method was utilized to calculate the specific surface areas using adsorption date in a relative pressure range from 0.05 to 0.95. The pore volumes and pore size distributions were derived from the desorption branches of the isotherms using the Barrett–Joyner–Halanda (BJH). The zeta potential of nanoparticles was measured by zeta potential (Nanotrac wave, Microtrac) at 25 °C with DI H2O as the solvent. pH-responsive release property of the drug was detected by UV-vis fluorescence spectrophotometer (LS50B, PerkinElmer) at 480 nm. Confocal laser scanning microscopy (CLSM, LSM 510Metanlo, Zeiss) was used to detect the distribution of the fluorescence-drug within the cells. The result of drug targeting delivery was conformed using the flow cytometry (Becton Dickinson).3 Results and discussion
3.1 The characterization of LA-BSA functionalized MHAPNs systems
The general procedure to construct the pH-responsive nanocarriers based on BSA-capped mesoporous hydroxyapatite nanoparticles (MHAPNs) was shown in Fig. 1. The MHAPNS was modified by amine group (NH2-MHAPNs) and followingly by 4-carboxyphenylboronic acid (CBA-MHAPNs). Then, after loading the model drugs into the CBA functionalized MHAPNs, the pore was blocked by the anchored LA-BSA via an esterification reaction. Thereafter, along with the breakage of cycle ester bonds linkages between LA-BSA and MHAPNs at acidic environments, the loaded drugs would be released to specific cells. | ||
| Fig. 1 Schematic illustration of intracellular pH-responsive system based on LA-BSA endcapped MHAPNs for cell targeting drug delivery. | ||
Firstly, the MHAPNs were synthesized employing the reported surfactant template method. From Fig. 2a, it can be seen that the prepared MHAPNs were uniform rod-like nanoparticles with a mean width of 50 nm and the length of 80 nm. In the highly magnified TEM image (Fig. 2a, inset), a highly ordered mesoporous network with an average pore size of 5 nm was clearly observed, which is the characteristic of MHAPNs. Also, the wide-angle XRD patterns of MHAPNs are shown in Fig. 2d, the three strong characteristic diffractions peaks (002, 211, 300) at 2θ = 25.8°, 31.8° and 32.9° were observed which can be indexed as pure hexagonal phase and coincide well with the standard date of Ca10(PO4)6(OH)2 (JCPDS 09-0432). Furthermore, from N2 adsorption–desorption isotherms studies (Fig. 3C), the BET isotherms of MHAPNs exhibited a typical Type IV isotherm cycle for mesoporous materials under the BDDT (Brunauer–Deming–Deming–Teller) system with a typical H1 hysteresis loop according to an IUPAC classification46 and a well-defined step at approximately P/P0 = 0.80–0.98. The curve again indicated the properties of typical mesoporous materials with a specific surface area of 183.46 m2 g−1, and average pore diameter of 5.06 nm with a narrow pore distribution, which was in agreement with the TEM observation.
 | ||
| Fig. 2 TEM images of MHAPNs (a) and LA-BSA-CBA-MHAPNs (b) (the inset shows the high magnification image). EDX spectrum of LA-BSA-CBA-MHAPNs (c), and XRD patterns of MHAPNs (d-black) and LA-BSA-CBA-MHAPNs (d-red). | ||
 | ||
| Fig. 3 (A) FTIR spectra of (a) MHAPNs, (b) NH2-MHAPNs, (c) CBA-MHAPNs, (d) LA-BSA-CBA-MHAPNs. (B) Zeta potential value of the each step modified MHAPNs measured in deionized water. (C) Nitrogen adsorption–desorption isotherms and (D) BJH pore size distributions for MHAPNs, NH2-MHAPNs, CBA-MHAPNs and LA-BSA-CBA-MHAPNs. | ||
Subsequently, the surface modification by amino group (NH2-MHAPNs) and 4-carboxyphenylboronic acid (CBA-MHAPNs) were performed. The successful synthesis of these two intermediates was confirmed by FTIR spectra (Fig. 3A). The band appeared at 1654 and 1563 cm−1 attributed to the N–H stretching vibrations and bending vibrations of NH2-MHAPNs (Fig. 3A-b), well showing that the amine groups have been grafted onto the surface of MHAPNs. While in Fig. 3A-c, the new bands at 1293 cm−1 belonged to C–H stretching vibrations of benzene, and absorption intensity of 1650 cm−1 became weaken, suggesting that parts of amine groups have been replaced by CBA molecules.
Finally, by esterification reaction between hydroxy groups of LA and boronic acid groups of CBA-MHAPNs, the pH-responsive MHAPNs nanocarrier with end-capped LA-BSA was constructed. As shown in Fig. 2b–d, the rod-like nanoparticle in the TEM image (Fig. 2b) and no other peaks shift in comparison of the unmodified MHAPNS in wide-angle XRD spectrum (Fig. 2d), demonstrated that the anchored LA-BSA had no effect on neither the morphology nor the typical hexagonal phase of the MHAPNs. Also from the highly magnified TEM image (Fig. 2b, inset), the blurry pore structure and the border around the MHAPNs, indicated the modification of LA-BSA onto the surface of MHAPNs. This was more clearly confirmed by FTIR spectra (Fig. 3A). After LA-BSA was anchored onto the surface of MHAPNs, the two appeared absorption peaks at 3430 cm−1 and 2926 cm−1 were due to the fact that the LA-BSA molecule had a relatively strong C–H skeleton vibration and absorption peak of COOH. Moreover, LA-BSA functionalized process on MHAPNs was tracked by zeta potential measurements in deionized water at each step (Fig. 3B). We can see that the potential value of the bare MHAPNs increased from −25.53 mV to +15.56 mV when amine groups were grafted onto the surface, since the presence of the amine groups on the surface which can be protonated. Then, when the NH2 on the surface of MHAPNs was partly esterified by CBA, the potential value of CBA-MHAPNs decreased to +5.56 mV, since the presence the boronic acid on the surface. Finally, after anchoring the LA-BSA onto the surface, the zeta potential value went down further to −12.86 mV. It is no doubt that the varied surface charge property in each step suggest the successful conjugating the functional groups onto the surface of MHAPNs. In addition, it should be mentioned that the N2 adsorption–desorption isotherms studies showed that after the capping of LA-BSA molecules, both the surface of the LA-BSA-CBA-MHAPNs and the pore volume reduced significantly (11.43 m2 g−1 and 0.0154 cm2 g−1 respectively, as shown in Table 1).
| Samples | SBET (m2 g−1) | Vp (cm3 g−1) | BJH (nm) |
|---|---|---|---|
| MHAPNs | 183.46 | 0.2689 | 5.06 |
| NH2-MHAPNs | 172.56 | 0.2386 | 4.88 |
| CBA-MHAPNs | 168.43 | 0.2193 | 4.60 |
| LA-BSA-CBA-MHAPNs | 11.43 | 0.0154 | — |
3.2 Biocompatibility of nanoparticles
Safety is of utmost importance when nanoparticles are used for biomedical applications. Although both hydroxyapatite and BSA have been reported to exhibit high biocompatibility, the cytotoxicity of newly synthesized materials in this study must be specified. To investigate the cell biocompatibility of LA-BSA-CBA-MHAPNs, an MTT assay was performed. The mechanism of MTT assay is based on the mitochondrial succinodehydrogenase within viable cells forming blue formazan crystals. It thus indirectly indicates the viability of cells. Considering the fact that both MHAPNs and BSA exhibit low- or non-cytotoxicity in the drug carrier, LA-BSA-CBA-MHAPNs showed a negligible effect on HepG2 cells (Fig. 4). However, cell viability decreased after incubation with both LA-BSA-CBA-MHAPNs@DOX and free DOX, since once the nanoparticles were engulfed and enter the cells and the loaded DOX was released leading to the decrease of the cell viability. Moreover, LA-BSA-CBA-MHAPNs@DOX showed remarkably higher anticancer efficiency than that of free DOX. The reason is that LA molecule bearing a galactose group is a specific ligand for the asialoglycoprotein receptor (ASGP-R) on the membrane of HepG2 cells.47,48 In general, LA-BSA-CBA-MHAPNs@DOX enhanced intracellular uptake and followed drug release resulting in enhanced killing efficacy as well as improved targeting specificity in comparison of conventional chemotherapy using DOX only. Thus, the MTT result demonstrated that the LA-BSA-CBA-MHAPNs@DOX could serve as a pH-responsive controlled release system for enhanced cancer targeted drug delivery.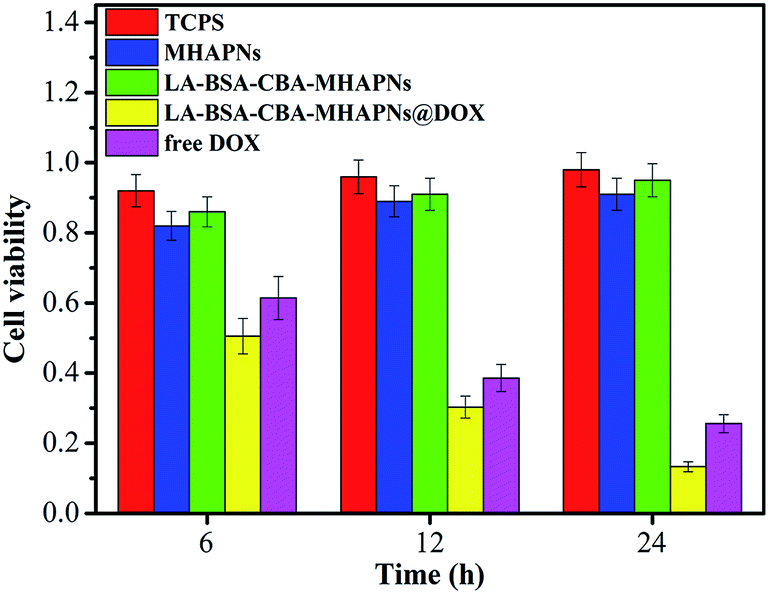 | ||
| Fig. 4 In vitro viability of HepG2 cells in the presence of TCPS (control group), MHAPNs, LA-BSA-CBA-MHAPNs, LA-BSA-CBA-MHAPNs@DOX and free DOX for 6 h, 12 h and 24 h, respectively. | ||
3.3 In vitro drug loading and pH-responsive release properties
To investigate the pH-responsive gating behavior of the system, time-dependent release of DOX from LA-BSA-CBA-MHAPNs@DOX system was carried out in buffered solutions at pH 7.4, 6.5, and 5.0, mimicking the physiological pH in normal tissue and blood, the tumor extracellular environment, and subcellular endosome, respectively.22 As shown in Fig. 5A, the release kinetics at different pH value are similar. However, at physiological medium (pH 7.4), about 8.0% of DOX was released out from LA-BSA-CBA-MSN@DOX within 5 h, this might be due to the partial physical degradation of BSA. However, around 34% and 53% of DOX were released out from LA-BSA-CBA-MHAPNs system when the pH value decreased to 6.5 and 5.0, respectively. The release rate of DOX increased apparently with the decrease of the pH value, which was attributed to the fact that cyclic ester bonds between CBA and LA-BSA molecules were stable at normal physiological conditions (pH 7.4), leading to stable end-capped MHAPNs without DOX release. When it was exposed in acidic environment (pH 6.5 or 5.0), the cyclic ester bonds could be easily hydrolyzed and allowed LA-BSA molecules to escape, resulting in the DOX release. Definitely, a stronger acidic environment (pH 5.0) will lead to a faster hydrolysis of the ester bonds than that of at pH 6.5 and shows a faster release rate of DOX. The result in Fig. 5A well indicates that BSA demonstrates a good end-capping efficiency to MHAPNs, and LA-BSA-CBA-MHAPNs@DOX is a highly pH sensitive system. To further investigate the rapid pH responsive drug release of LA-BSA-CBA-MHAPNs@DOX, the pH value of incubation solution was adjusted to 5.0 when LA-BSA-CBA-MHAPNs@DOX were incubated with a medium of pH 7.4. The result shows only 8% of DOX was released in the initial 5 h (pH 7.4), whereas the cumulative release quantity of DOX reached 55% after adjusting pH to 5.0 in the next 5 h. The released DOX could be clearly visualized from the change of the color of the solution. The phenomenon further demonstrated that LA-BSA-CBA-MHAPNs@DOX system showed a rapid responsive to pH. | ||
| Fig. 5 Cumulative release profiles of DOX. (A) Controlled release of DOX from LA-BSA-CBA-MHAPNs@DOX systems in PBS at different pH values. (B) The accelerated release of DOX from LA-BSA-CBA-MHAPNs@DOX systems when the pH value is adjusted from 7.4 in the first 5 h to 5.0. | ||
3.4 Intracellular drug delivery
To investigate cell uptake and intracellular distribution of MHAPNs@FITC (no capping) and LA-BSA-CBA-MHAPNs@FITC nanocomposites, FITC was employed as a model drug for intracellular tracing. The released FITC was observed with confocal laser scanning microscopy (CLSM) after incubation with HepG2 cells. After incubation with MHAPNs@FITC and LA-BSA-CBA-MHAPNs@FITC, cells displayed well-spreading morphology, indicating their good cytocompatibilities. As shown in Fig. 6, the green fluorescence from internalized FITC illustrated in the confocal image clearly indicates that drug molecules are released into the HepG2 cells by both MHAPNs and LA-BSA-CBA-MHAPNs carriers. Furthermore, from the CLSM images (Fig. 6), one can see that after 6 h incubation, there was only a small amount of fluorescent particles found in the cytoplasm of HepG2 cells. While, with prolonged time, the number of fluorescent particles in cytoplasm increased significantly after 12 h and 24 h incubation. The result illustrates the MHAPNs and LA-BSA-CBA-MHAPNs could be effectively internalized into living cells and endocytosis efficiency increased along with incubation time. However, LA-BSA-CBA-MHAPNs@FITC displayed markly higher cell uptake efficiency than that of MHAPNs for 6 h, 12 h, and 24 h incubation, respectively. In comparison of that of MHAPNs, the higher cell uptake of LA-BSA-CBA-MHAPNs@FITC could be attributed to the receptor-mediated endocytosis.47–49 The ASGP-R on the membrane of HepG2 cells could specifically identify LA, resulting in an efficient endocytosis. To demonstrate the enhanced specific uptake capability of LA-BSA-CBA-MHAPNs@FITC, the endocytosis efficiency was quantified by flow cytometry. LA-BSA-CBA-MHAPNs@FITC and MHAPNs@FITC were incubated with HepG2 cells for 6 h, 12 h and 24 h, respectively. LA-BSA-CBA-MHAPNs@FITC shows excellent endocytosis efficiencies which are about 2.3, 2.1 and 2.3 times higher than that of MHAPNs after incubation for 6 h, 12 h and 24 h, respectively (Fig. 7a). The mechanism is that a large amount of specific ligand-asialoglycoprotein receptor (ASGP-R) of lactobionic acid exists on the membrane of HepG2 cells, which the galactose groups on LA-BSA-CBA-MHAPNs can specifically recognize. The ASGP receptor (ASGP-R) is a glycoprotein which can specifically recognize galactosyl ligands via a multivalent interaction, and ASGP-R expresses exclusively in HepG2 cells.50 Therefore, these results confirm that, taking advantage of the galactose conjugation, the LA-BSA-CBA-MHAPNs is able to be more effective for cellular uptake than that of MHAPNs. | ||
| Fig. 6 Cell uptake and intracellular distribution of nanoparticles. Representative CLSM images of MHAPNs@FITC (a1–a3) and LA-BSA-CBA-MHAPNs@FITC (b1–b3), endocytosed by HepG2 cells after culture for 6 h, 12 h, and 24 h, respectively. | ||
 | ||
| Fig. 7 (a) Quantification of endocytosis of MHAPNs@FITC and LA-BSA-CBA-MHAPNs@FITC and by HepG2 cells; (b) flow cytometry analysis of the specific endocytosis of LA-BSA-CBA-MHAPNs@FITC by HepG2 cells and endothelial cells. | ||
To confirm the potential of LA-BSA-CBA-MHAPNs to be a targeted drug delivery carrier, we further demonstrated the endocytosis and intracellular delivery of nanoparticles by HepG2 cells and HUVECs. As depicted in Fig. 7b, the endocytosis number of HepG2 cells that internalized LA-BSA-CBA-MHAPNs is markedly higher than that of HUVECs. It was 2.1, 2.0 and 2.2 times higher after incubation for 6 h, 12 h and 24 h, respectively. The result again indicates LA on the surface MHAPNs plays an important role in enhancing endocytosis via a multivalent interaction with ASGP receptor on the surface of HepG2 cells. It further verified that LA functioned MHAPNs had the capacity to target HepG2 cells. The targeting mechanism was consistent with the MTT assay and CLSM analysis. Taken together, we confirmed our research that LA-BSA capped MHAPNs could serve as nanocarriers in a pH-responsive controlled release system for targeted drug delivery.
4 Conclusions
In summary, we have successfully designed and synthesized a targeted release nanocarriers based on MHAPNs for intracellular pH-responsive delivery of anticancer drugs by using LA-BSA molecules as pH-responsive valve. LA-BSA-CBA-MHAPNs system exhibited good drug loading capacity as well as enhanced drug release efficiency and targeting ability to cancer cells. The release of the loaded drugs at normal physiological conditions (pH 7.4) was inhibited, but such release rate could be accelerated dramatically at acidic environments (pH 5.0) by the cleavage of pore-blocking LA-BSA caps. More important, the galactose group of LA-BSA-CBA-MHAPNs can specifically recognize the asialoglycoprotein receptor (ASGP-R) on the membrane of HepG2 cells via a multivalent interaction, thus the system enhanced intracellular uptake and targeting specificity. Overall, in view of the excellent biocompatibility, nontoxicity, targeted cellular uptake properties, and efficient intracellular pH-responsive, the reported system shows great potential for the biomedical application in vivo controlled drug release in the future.Acknowledgements
This work was financially supported by Shandong Province Postdoctoral Foundation of China (201101003), China Postdoctoral Science Foundation (2013M541372), Fundamental Research Funds for the Central Universities (HIT. NSRIF2015047), and Weihai Science and Technology Development Plan project (2013GNS028) from Weihai city, Shandong province of China.References
- R. Liu, Y. Zhan, X. Zhao, A. Agarwal, L. J. Mueller and P. Y. Feng, J. Am. Chem. Soc., 2010, 132, 1500–1501 CrossRef CAS PubMed.
- H. P. Rim, K. H. Min, H. J. Lee, S. Y. Jeong and S. C. Lee, Angew. Chem., Int. Ed., 2011, 50, 8853–8857 CrossRef CAS PubMed.
- P. Pakawanit, S. Ananta, T. K. Yun, J. Y. Bae, W. Jang, H. Byun and J. H. Kim, RSC Adv., 2014, 4, 39287–39296 RSC.
- Z. Y. Zhou, S. M. Zhu and D. Zhang, J. Mater. Chem., 2007, 17, 2428–2433 RSC.
- Y. Qiu and K. Park, Adv. Drug Delivery Rev., 2001, 53, 321–339 CrossRef CAS.
- C. H. Lee, S. H. Cheng, I. P. Huang, J. S. Sours, C. S. Yang, C. Y. Mou and L. W. Lo, Angew. Chem., Int. Ed., 2010, 49, 8214–8219 CrossRef CAS PubMed.
- X. Shi, Y. Zhen, G. Wang, Q. Lin and J. Fan, RSC Adv., 2014, 4, 47056–47065 RSC.
- R. Casasus, E. Climent, D. Marcos, F. Sancenon, J. Soto, J. Cano and E. Ruiz, J. Am. Chem. Soc., 2008, 130, 1903–1917 CrossRef CAS PubMed.
- Y. Zhu, J. Shi, W. Shen, X. Dong, J. Feng, M. Ruan and Y. Li, Angew. Chem., Int. Ed., 2005, 44, 5083–5087 CrossRef CAS PubMed.
- D. Feng, J. Shi, X. Wang, L. Zhan and S. Cao, RSC Adv., 2013, 3, 24795–24982 Search PubMed.
- C. R. Thomas, D. P. Ferris, J. H. Lee, E. Choi, M. H. Cho, J. S. Shin, J. Cheon and J. I. Zink, J. Am. Chem. Soc., 2010, 132, 10623–10625 CrossRef CAS PubMed.
- B. T. Nguyen, K. C. Leung, M. Liong, Y. Liu, J. F. Stoddart and J. I. Zink, Adv. Funct. Mater., 2007, 17, 2101–2110 CrossRef.
- C. Y. Park, K. H. Lee and C. H. Kim, Angew. Chem., Int. Ed., 2009, 48, 1275–1278 CrossRef CAS PubMed.
- D. P. Ferris, Y. L. Zhao, N. M. Khashab, H. A. Khatib, J. F. Stoddart and J. I. Zink, J. Am. Chem. Soc., 2009, 131, 1686–1688 CrossRef CAS PubMed.
- Z. Luo, K. Cai, Y. Hu, Z. Li, L. Peng, D. Lin and W. Yang, Angew. Chem., Int. Ed., 2011, 50, 640–643 CrossRef CAS PubMed.
- D. Liu, X. Zhao, T. Wu and P. Y. Feng, J. Am. Chem. Soc., 2008, 130, 14418–14419 CrossRef PubMed.
- B. H. Kim, H. Matsuda, H. S. Zhou and I. Honma, Adv. Mater., 2006, 18, 3083–3088 CrossRef.
- J. Xuan, M. Pelletier, H. Xia and Y. Zhao, Macromol. Chem. Phys., 2008, 212, 498–506 CrossRef.
- A. Schlossbayer, J. Kecht and T. Bein, Angew. Chem., Int. Ed., 2009, 48, 3092–3095 CrossRef PubMed.
- K. Radhakrishman, J. Tripathy, D. P. Gnanadhas, D. Chakravortty and A. M. Raichur, RSC Adv., 2014, 4, 45961–45968 RSC.
- A. J. Harnoy, I. Rosenbaum, E. Tirosh, Y. Ebenstein, R. Shaharabani, R. Beck and R. J. Amir, J. Am. Chem. Soc., 2014, 136, 7531–7534 CrossRef CAS PubMed.
- J. Su, F. Chen, V. L. Cryns and P. B. Messersmith, J. Am. Chem. Soc., 2011, 133, 11850–11853 CrossRef CAS PubMed.
- K. Engin, D. B. Leeper, J. R. Cater, A. J. Thistlethwaite, L. Tupchong and I. D. Mcfarlane, Int. J. Hyperthermia, 1995, 11, 211–216 CrossRef CAS PubMed.
- S. L. Schmid, R. Fuchs, P. Male and I. Mellman, Cell, 1988, 52, 73–83 CrossRef CAS.
- H. Zhen and S. Chen, RSC Adv., 2012, 2, 4421–4429 RSC.
- N. Singh, A. Karambelkar, L. Gu, K. Lin, J. S. Miller, C. S. Chen, M. J. Sailor and S. N. Bhatia, J. Am. Chem. Soc., 2011, 133, 19582–19585 CrossRef CAS PubMed.
- X. Huang, N. Hauptmann, D. Appelhans, P. Formanek, S. Frank, S. Kaskel, A. Temme and B. Voit, Small, 2012, 8, 3579–3583 CrossRef CAS PubMed.
- B. H. Kim, S. Kim, C. Park, H. Lee, H. J. Park and C. Kim, Adv. Mater., 2010, 22, 4280–4283 CrossRef PubMed.
- B. A. Rammohan, L. Tayal, A. Kumar, S. Sivakumar and A. Sharma, RSC Adv., 2013, 3, 2008–2016 RSC.
- R. K. Singh, T. H. Kim, J. J. Kim, E. J. Lee, J. C. Knowles and H. W. Kim, RSC Adv., 2013, 3, 8692–8704 RSC.
- Y. Lin, Z. Li, Z. Chen, J. Ren and X. Qu, Biomaterials, 2013, 34, 2600–2610 CrossRef CAS PubMed.
- N. K. Mal, M. Masahiro and Y. Tanaka, Nature, 2003, 421, 350–353 CrossRef CAS PubMed.
- S. S. Sandra, C. Montserrat, I. B. Isable and V. R. Maria, J. Mater. Chem. B, 2013, 1, 1595–1606 RSC.
- M. Li, Y. Wang, Q. Liu, Q. Li, Y. Cheng, Y. Zheng, T. Xi and S. Wei, J. Mater. Chem. B, 2013, 1, 475–484 RSC.
- Y. H. Zhao and J. Ma, Microporous Mesoporous Mater., 2005, 87, 110–117 CrossRef CAS PubMed.
- J. Zhang, Q. Wang and A. Wang, Acta Biomater., 2010, 6, 445–454 CrossRef CAS PubMed.
- G. Bharath, A. J. Kumar, K. Karthick, D. Mangalaraj, C. Viswanathan and N. Ponpandian, RSC Adv., 2014, 4, 37446–37457 RSC.
- Z. Li, Z. Liu, M. Yin, X. Yang, Q. Yuan, J. Ren and X. Qu, Biomacromolecules, 2012, 13, 4257–4263 CAS.
- D. Li, J. He, W. Cheng, Y. Wu, Z. Hu, H. Tian and Y. Huang, J. Mater. Chem. B, 2014, 2, 6089–6096 RSC.
- R. K. Singh, T. H. Kim, K. D. Patel, J. J. Kim and H. W. Kim, J. Mater. Chem. B, 2014, 2, 2039–2050 RSC.
- J. S. Son, M. Appleford, J. L. Ong, J. C. Wenke, J. M. Kim, S. H. Choi and D. S. Oh, J. Controlled Release, 2011, 153, 133–140 CrossRef CAS PubMed.
- A. Bernardos, E. Aznar, M. D. Marcos, M. R. Martinez, F. Sancenon, J. Soto, J. M. Barat and P. Amoros, Angew. Chem., Int. Ed., 2009, 48, 5884–5887 CrossRef CAS PubMed.
- Z. Luo, K. Cai, Y. Hu, B. Zhang and D. Xu, Adv. Healthcare Mater., 2012, 1, 321–325 CrossRef CAS PubMed.
- Y. Zhao, B. G. Trewyn, I. I. Slowing and V. S. Lin, J. Am. Chem. Soc., 2009, 131, 8398–8400 CrossRef CAS PubMed.
- E. Aznar, M. D. Marcos, R. Martinez-Manez, F. Sancenon, J. Soto, P. Amoros and C. Guillem, J. Am. Chem. Soc., 2009, 131, 6833–6843 CrossRef CAS PubMed.
- P. Nabakumar and I. Tokoko, Langmuir, 2012, 28, 14018–14027 CrossRef PubMed.
- I. I. Slowing, B. G. Trewyn and V. S. Lin, J. Am. Chem. Soc., 2006, 128, 14792–14793 CrossRef CAS PubMed.
- L. Du, S. Liao, H. A. Khatib, J. F. Stoddart and J. L. Zink, J. Am. Chem. Soc., 2009, 131, 15136–15142 CrossRef CAS PubMed.
- E. M. Kim, H. J. Jeong, S. L. Kim, J. W. Nah, H. S. Bom, I. K. Park and C. S. Cho, Nucl. Med. Biol., 2006, 33, 529–534 CrossRef CAS PubMed.
- B. C. H. Lai, C. Y. Lin, H. T. Wu, H. S. Chan, Y. J. Chuang, C. T. Chen and C. C. Lin, Adv. Funct. Mater., 2010, 20, 3948–3958 CrossRef.
| This journal is © The Royal Society of Chemistry 2015 |