
 Open Access Article
Open Access Article This Open Access Article is licensed under a
This Open Access Article is licensed under a Creative Commons Attribution 3.0 Unported Licence
Phenothiazine-functionalized redox polymers for a new cathode-active material†
Ali A.
Golriz
ab,
Takeo
Suga
*c,
Hiroyuki
Nishide
*d,
Rüdiger
Berger
a and
Jochen S.
Gutmann
*be
aMax Planck Institute for Polymer Research, Ackermannweg 10, D-55128 Mainz, Germany. E-mail: berger@mpip-mainz.mpg.de; Fax: +49-6131-379-100; Tel: +49-6131-379-114
bDepartment of Chemistry and Center for Nanointegration Duisburg-Essen (CENIDE), University Duisburg-Essen, Universitätsstr. 5, D-45117 Essen, Germany. E-mail: jochen.gutmann@uni-due.de; Fax: +49-201-183-4934; Tel: +49-201-183-2566
cWaseda Institute for Advanced Study, Waseda University, Tokyo 169-8555, Japan. E-mail: takeosuga@toki.waseda.jp
dDepartment of Applied Chemistry, Waseda University, Tokyo, 169-8555, Japan. E-mail: nishide@waseda.jp; Fax: +81-3-3209-5522; Tel: +81-3-3200-2669
eDeutsches Textilforschungszentrum Nord-West e.V., Adlerstr. 1, D-47798 Krefeld, Germany
First published on 24th February 2015
Abstract
Redox-active, phenothiazine-functionalized polymers were synthesized and employed as a promising cathode-active material (∼3.7 V vs. Li, 77 Ah kg−1) in a rechargeable battery. The longer spacer between phenothiazine and the polymer backbone contributed to the stability of the formed radical cations, resulting in decelerated self-discharge and improved cycle performance.
Electro-active polymers bearing redox-active pendant groups such as triphenylamine, carbazole, and nitroxide, etc. have attracted renewed attention in energy-storage and opto-/electronics applications.1,2 The phenothiazine unit, a well-known hetero-tricyclic structure in methylene blue, photo-sensitizers,3 photo-polymerization catalysts,4 and pharmacological agents,5 has been also proposed as a key constituent for the hole-transporting layer in polymer light emitting diodes (PLED),6 for electrocatalysts in biosensors,7 and for electrochromic materials in electronic displays.8 At molecular basis, such superior electronic characteristics lies in the redox chemistry of phenothiazine units.9,10 The oxidation of the neutral phenothiazine leads to the formation of persistent radical cation species, and their π–π stacking results in enhanced electric conductivity.11 Remarkable is the stability of both neutral and cationic states (>50 h). We have successfully demonstrated that a thin film of poly-3-vinylbenzylphenothaizine can be used as a bias-responsive bistable media for data storage.12,13 On a spin-coated layer of phenothiazine-based polymers, nano-patterns with enhanced conductivity were recorded via conductive scanning force microscopy,14 and conductive states (cations) were stored for several days. Furthermore, grafted polymer brushes for phenothiazine polymers were developed to obtain redox-active surfaces with increased nanowear stability.15,16
Such long-term durability of phenothiazine radical cations at a solid-state (without electrolyte) has evoked renewed attention in their potential electrochemical applications, where the redox process can be supported/stabilized in presence of electrolytes. Recent progress in utilizing redox polymers for electrode-active materials in a rechargeable battery has been demonstrated by Nishide.17,18 Nitroxide-functionalized polymers were employed as cathode-active material to give the cell voltage of 3.5 V vs. Li/Li+, relatively high charge capacity (110 mA h g−1), high power-rate performance (rapid charging), and long cycle life (>1000 cycles). Other redox-active organic groups such as benzoquinones,19 heteroaromatic derivatives20 have been re-examined in terms of charge-storage capability. Phenothiazine is a promising candidate for a new cathode-active material to show more positive oxidation potential, which leads to higher cell voltage. Enhanced conductivity based on π–π stacking of closely immobilized phenothiazine side groups promises associated conductivity besides on redox conduction. Phenothiazine-based polymers can be prepared in a facile conventional radical polymerization, compared with non-trivial synthesis of radical polymers, which often requires the protecting groups or precursors for the polymerization steps.21,22 There are a few previous reports dealing with electrochemical properties of phenothiazine-based polymers,23 but here we report synthesis and electrochemical properties of phenothiazine polymers in view of their application to cathode-active materials in a lithium-ion battery. In particular, we synthesize phenothiazine-functionalized polymers with different alkyl spacer lengths, poly(10-(4-vinylbenzyl)-10H-phenothiazine) (PVBPT) and poly(10-(3-(4-vinylphenyl)propyl)-10H-phenothiazine) (PVPPP), and elucidate whether the spacer plays a key role for tuning the redox stability, which closely related to self-discharge behaviour and cycle performance of the battery.
Phenothiazine-functionalized polymers, PVBPT (Mn = 273![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) 000, PDI = 2.32) and PVPPP (Mn = 25800, PDI = 1.96) were synthesized via conventional radical polymerization from the corresponding monomers in anisole or THF using AIBN (1 mol%) as an initiator (Fig. 1). Detailed synthetic procedures were shown in the ESI.† The obtained polymers were spin-coated on an indium tin oxide (ITO) electrode (thickness ∼100–150 nm) and used for electrochemical characterizations.
000, PDI = 2.32) and PVPPP (Mn = 25800, PDI = 1.96) were synthesized via conventional radical polymerization from the corresponding monomers in anisole or THF using AIBN (1 mol%) as an initiator (Fig. 1). Detailed synthetic procedures were shown in the ESI.† The obtained polymers were spin-coated on an indium tin oxide (ITO) electrode (thickness ∼100–150 nm) and used for electrochemical characterizations.
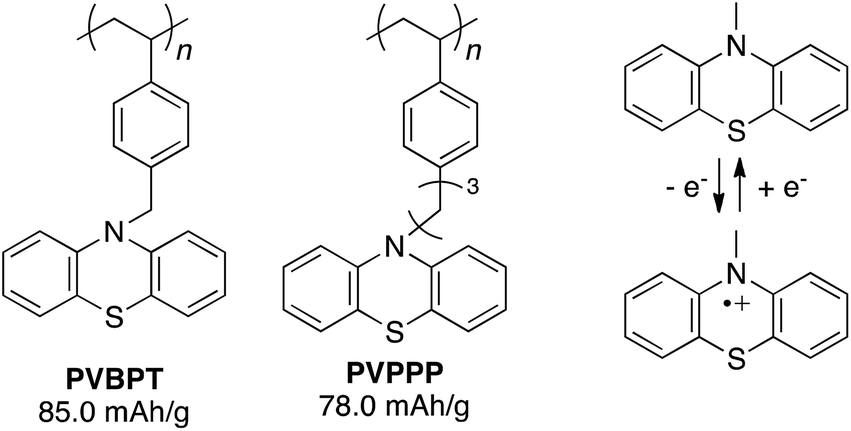 | ||
| Fig. 1 Chemical structure of phenothiazine-functionalized polymers and the redox couple between phenothiazine and its radical cation. | ||
Cyclic voltammograms for PVBPT and PVPPP films on ITO exhibited reversible redox waves at E0 = 0.87 and 0.84 V vs. Ag/AgCl, respectively (Fig. 2a and b). Formation of the radical cation species was examined via electron spin resonance (ESR) spectroscopy combined with CV. The oxidation of PVPPP at 1.1 V formed an ESR signal at g = 2.0049, (2.0052 reported for benzylphenothiazine),24 which suggested the free electron is delocalized through the aromatic ring systems, and appeared as carbon-centered radicals. The broadening of the signal was explained by the high local concentration of radical species along the backbone. The ESR signal disappeared after applying potential at 0.5 V, which demonstrated the reversible reduction of the radical cation to the neutral state (Fig. 2 inset).
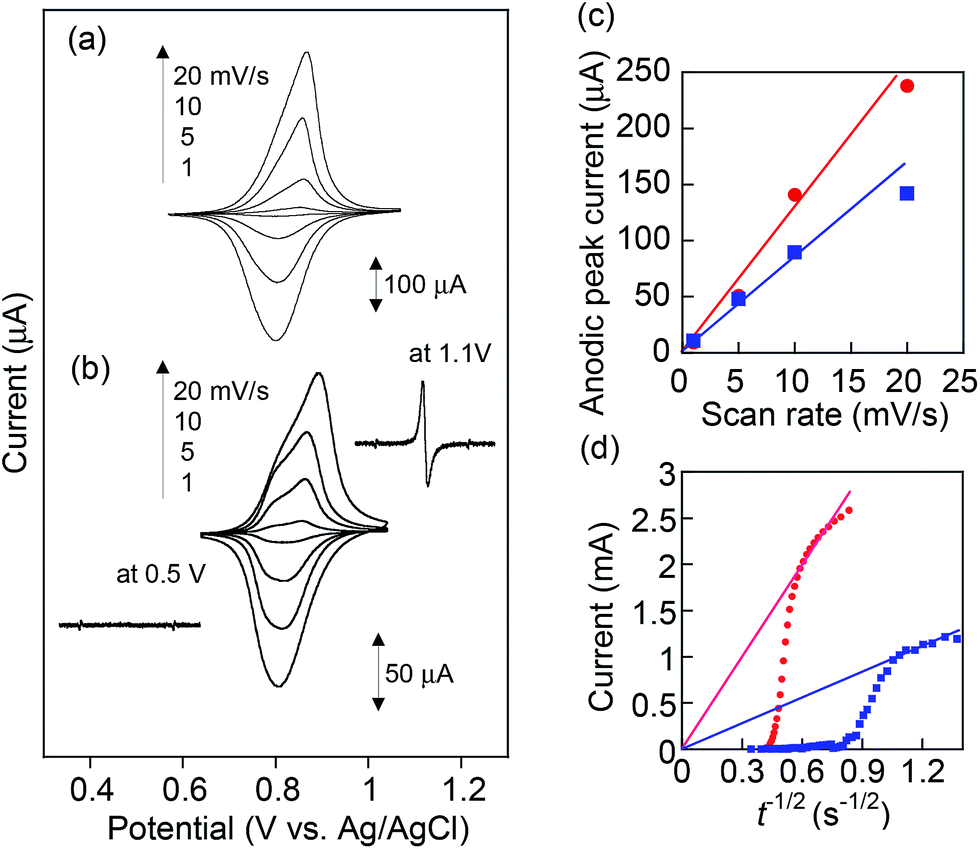 | ||
| Fig. 2 Cyclic voltammograms of (a) PVBPT and (b) PVPPP spin-coated on indium tin oxide (ITO) as a working electrode. Inset: in situ electrolytic ESR spectra under the applied potentials. (c) Anodic peak currents vs. scan rates and (d) Cottrell plots for PVBPT (red) and PVPPP (blue). | ||
The separation between oxidation and reduction peaks, ΔEp, for PVBPT slightly increased from 48 to 67 mV at increasing scan rates from 1 to 20 mV s−1, and the linear plots of the measured peak currents against the scan rates for the anodic (or cathodic) scans indicated the reversible redox characteristics for the surface-confined redox species (Fig. 2c). PVPPP exhibited an increased peak-to-peak separations at scan rates ≥ 10 mV s−1. An anodic peak current obtained at a scan rate of 20 mV s−1 deviated from a linear correlation between peak currents vs. scan rates, which suggested slower redox process for PVPPP film.
The electron transfer kinetics through the film was further investigated by chronoamperometry (CA) measurements. The apparent diffusion coefficients, Dapp, of electron transfer through the PVBPT and PVPPP films were estimated from the Cottrell plots (Fig. 2d) to be 1.5 × 10−9 and 1.0 × 10−10 (cm2 s−1), respectively, based on an equation of i = nFDapp1/2C*/π1/2t1/2.25 Both values are in good agreement with the reported values from the literature.24 From the calculated values for Dapp we estimated the electron exchange rate kex using the Dahms–Ruff equation: kex = 4Dapp/(πδ2C*redox).26 The estimation requires the center-to-center distance δ of the redox moieties in the polymer chain (≈5 Å) and the concentration of the redox species at the electrode interface C*redox, which was calculated from the CA data. A faster electron self-exchange rate, kex for PVBPT was obtained to be 1.6 × 106 M−1 s−1 compared with that of PVPPP (4.9 × 104 M−1 s−1), which suggested long alkyl spacer group between phenothiazine and polymer backbone enabled more rotational freedom for the aromatic rings to get in the proper juxtaposition for electron exchange. In PVPPP, the redox moieties were separated from each other so that the interaction between them was aggravated and the rotation process involved additional activation. This results in a reduced the electron self-exchange rate.
The chemical stability of the oxidized state (radical cations) for PVBPT and PVPPP was studied by in situ electrochemical UV and ESR spectroscopy. The oxidation process was accompanied by reversible color change from colorless to dark red (Fig. 3b, inset). The first absorption maximum at 518 nm corresponded to the absorption bands for the phenothiazine radical cation, and the second absorption band at 859 nm corresponded to the formation of a dimer with a face-to-face orientation of the radical cation species (Fig. 3).27,28 The stronger absorption peak at 800–900 nm for oxidized PVBPT compared with oxidized PVPPP suggested facile dimer formation of phenothiazine cations due to the short spacer groups, which also supported the electron transfer kinetics. After oxidation, the bias was turned off, and the UV spectral change for both polymers was monitored (Fig. 3a and b). The absorption peaks at 518 nm decreased over time for PVBPT, and relative absorbance after 60 min was 60% (Fig. 3c). Re-oxidation of PVBPT exhibited the original peak intensity, which indicated the time decay was a reversible process. On the other hand, the absorption peak for PVPPP remained almost unchanged over time, which indicates that the long spacer group contributes the radical stability.
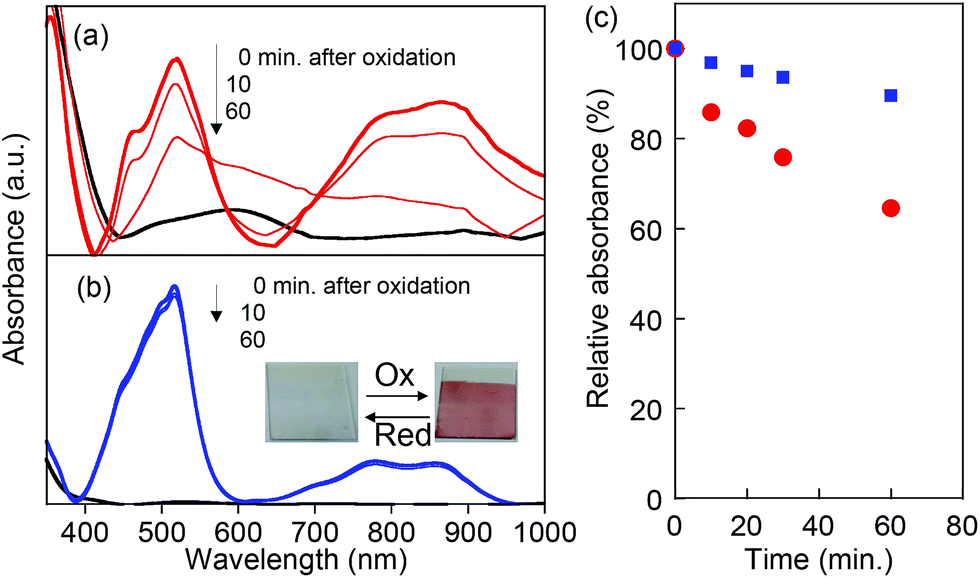 | ||
| Fig. 3 In situ electrochemical UV spectra of (a) PVBPT and (b) PVPPP on ITO electrode. A neutral phenothiazine form was shown in black, and the corresponding radical cations under the applied potentials at 1.1 V were shown in red and blue, respectively. (c) Time decay of the relative absorbance at 518 nm ascribed to the radical cations for PVBPT (red) and PVPPP (blue). | ||
To examine the applicability of both phenothiazine-based polymers as cathode-active materials, chronopotentiometry (CP) experiments were performed. Each polymer was combined with vapor-grown carbon fibers as conductive additives, and poly(vinylidene fluoride) as a binder, and coated on glassy carbon electrode or aluminum foil to prepare the composite electrode. PVPPP electrode exhibited a charging capacity of Qc = 75 Ah kg−1 and a discharging capacity of Qd = 72 Ah kg−1 at a C-rate of 10 C, which corresponded to 96% and 92% of the theoretical calculation (Fig. 4). The coulomb efficiency (ratio of Qd/Qc) was approximately 97% at all C-rates showing a quantitative redox process in both directions. The plateau voltages slightly increased from 0.85 to 0.93 V at increasing C-rate from 10 to 50 C, and the measured charge capacity decreased to 56 Ah kg−1 (ca. 25% decrease compared with the initial capacity) at 50 C. However, an almost quantitative charging and discharging was achieved up to 20 C-rates. On the other hand, Qc and Qd for PVBPT were 79 and 61 Ah kg−1, respectively, resulting in 77% coulomb efficiency. Such self-discharge phenomena can be explained by the lower stability of phenothiazine cation radical for PVBPT with a short spacer group.
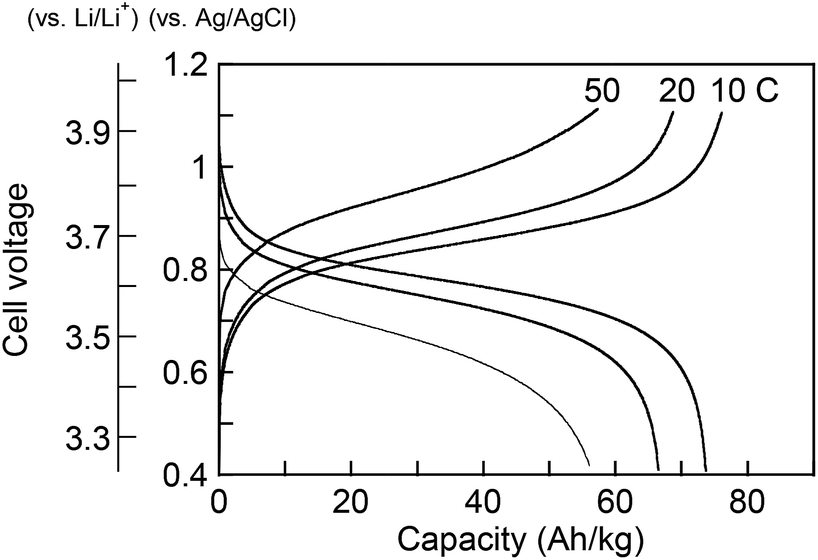 | ||
| Fig. 4 Charge–discharge curves of PVPPP composite electrodes on glassy carbon. | ||
To examine the long-term cycle performance of the polymers, PVBPT and PVPPP composite electrodes were installed as a cathode in a coin cell battery with lithium metal anode (Fig. 5). Both polymers demonstrated plateau voltages of around 3.7 V, which was advantageous for high voltage battery devices. The overall cell potential increased around 0.2 V compared to nitroxide-based polymer cathodes.17 The coin cell battery was tested by 500 cycles of sequential charging and discharging CP experiments (Fig. S1†). While after 500 cycles the capacity of PVPPP decreased 12% compared to the initial capacity, PVBPT showed a decrease of 27%. The increase in cycle stability can be attributed to the resonance stabilizing effect of the propylene spacer in PVPPP. The non-branched alkyl spacer stabilizes the radical cation species in the oxidized, coplanar state of phenothiazine.
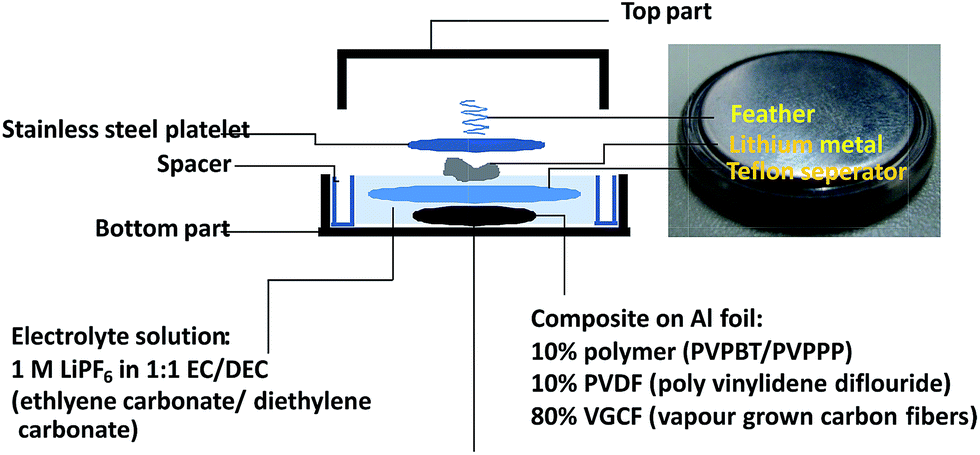 | ||
| Fig. 5 Schematic setup of a coin cell battery with PVBPT and PVPPP as cathode-active material. | ||
Conclusions
In summary, we could successfully integrate polymers with phenothiazine redox moieties as p-type cathode-active material in a lithium coin cell battery. We obtained a high cell voltage of ∼3.7 V by using a phenothiazine-based cathode. This value is promising for utilization in a polymer-based rechargeable battery. One important finding in our study is related to the effect of spacer between the phenothiazine moiety and the polymer backbone. The increase in spacer length decreased the electron self-exchange rate but simultaneously stabilized the radical cation formed by oxidation, resulting in decelerated self-discharge and improved cycle performance. Our findings offer new molecular design approaches to tune redox stability. Further optimization of cell configuration and electrolyte conditions will be discussed in future studies.Acknowledgements
This project was partially supported by the Strategic Japanese German 756 Cooperative Programme On Nanoelectronics (BE3286/2-1; 757 GU 771/6-1), The Japan Science and Technology Agency (JST), and the International Research Training Group 1404 “Self-organized Materials for Optoelectronics” (DFG). This project was partially supported by Grants-in Aid for Scientific Research (nos 24225003 and 25620179) from MEXT, Japan. T. S. acknowledges the MEXT/JST tenure-track program.Notes and references
- For reviews and book chapters: (a) H. Nishide and T. Suga, Electrochem. Soc. Interface, 2005, 14(4), 32 CAS; (b) K. Nakahara, K. Oyaizu and H. Nishide, Chem. Lett., 2011, 40, 222 CrossRef CAS; (c) P. Poizot and F. Dolhem, Energy Environ. Sci., 2011, 4, 2003 RSC; (d) T. Janoschka, M. D. Hager and U. S. Schubert, Adv. Mater., 2012, 24, 6397 CrossRef CAS PubMed.
- H. Nishide and K. Oyaizu, Science, 2008, 319, 737 CrossRef CAS PubMed.
- Z. Gomurashvili and J. V. Crivello, Macromolecules, 2002, 35, 2962 CrossRef CAS.
- N. J. Treat, H. Sprafke, J. W. Kramer, P. G. Clark, B. E. Barton, J. R. de Alaniz, B. P. Fors and C. J. Hawker, J. Am. Chem. Soc., 2014, 136, 16096 CrossRef CAS PubMed.
- C. Korth, B. C. H. May, F. E. Cohen and S. B. Prusiner, Proc. Natl. Acad. Sci. U. S. A., 2001, 98, 9836 CrossRef CAS PubMed.
- (a) S. K. Kim, J. H. Lee and D. H. Hwang, Synth. Met., 2005, 152, 201 CrossRef CAS PubMed; (b) D.-H. Hwang, S.-K. Kim, M.-J. Park, J.-H. Lee, B.-W. Koo, I.-N. Kang, S.-H. Kim and T. Zyung, Chem. Mater., 2004, 16, 1298 CrossRef CAS.
- E. Dominiquez, H. L. Lan, Y. Okamoto, P. D. Hale, T. A. Skotheim, L. Gorton and B. Hahnhagerdal, Biosens. Bioelectron., 1993, 8, 229 CrossRef.
- F. Fungo, S. A. Jenekhe and A. J. Bard, Chem. Mater., 2003, 15, 1264 CrossRef CAS.
- G. P. Brown and S. Aftergut, Nature, 1962, 193, 361 CrossRef CAS.
- J. P. Billon, Bull. Soc. Chim. Fr., 1960, 1784 CAS.
- (a) Z. Zhou, A. W. Franz, M. Hartmann, A. Seifert, T. J. J. Müller and W. R. Thiel, Chem. Mater., 2008, 20, 4986 CrossRef CAS; (b) Z. Zhou, A. W. Franz, S. Bay, B. Sarkar, A. Seifertm, P. Yang, A. Wagener, S. Ernst, M. Pagels, T. J. J. Müller and W. R. Thiel, Chem.–Asian J., 2010, 5, 2001 CrossRef CAS PubMed; (c) M. Sailer, A. W. Franz and T. J. J. Müller, Chem.–Eur. J., 2008, 14, 2602 CrossRef CAS PubMed; (d) T. J. J. Müller, A. W. Franz, C. S. Barkschat, M. Sailer, K. Meerholz, D. Müller, A. Colsmann and U. Lemmer, Macromol. Symp., 2010, 287, 1 CrossRef.
- A. A. Golriz, T. Kaule, J. Heller, M. B. Untch, P. Schattling, P. Theato, M. Toda, S. Yoshida, T. Ono, H. J. Butt, J. S. Gutmann and R. Berger, Nanoscale, 2011, 3, 5049 RSC.
- A. A. Golriz, T. Kaule, M. B. Untch, K. P. Kolman, R. Berger and J. S. Gutmann, ACS Appl. Mater. Interfaces, 2013, 5, 2485 CAS.
- R. Berger, H.-J.- Butt, M. Retschke and S. Weber, Macromol. Rapid Commun., 2009, 30, 1167 CrossRef CAS PubMed.
- R. Berger, Y. Cheng, R. Förch, B. Gotsmann, J. S. Gutmann, T. Pakula, U. Rietzler, W. Schärtl, M. Schmidt, A. Strack, J. Windeln and H.-J. Butt, Langmuir, 2007, 23, 3150 CrossRef CAS PubMed.
- S. A. Pihan, S. G. J. Emmerling, H.-J. Butt, J. S. Gutmann and R. Berger, Wear, 2011, 271, 2852 CrossRef CAS PubMed.
- H. Nishide, S. Iwasa, Y. J. Pu, T. Suga, K. Nakahara and M. Satoh, Electrochim. Acta, 2004, 50, 827 CrossRef CAS PubMed.
- T. Suga, H. Ohshiro, S. Sugita, K. Oyaizu and H. Nishide, Adv. Mater., 2009, 21, 1627 CrossRef CAS.
- W. Choi, D. Harada, K. Oyaizu and H. Nishide, J. Am. Chem. Soc., 2011, 133, 19839 CrossRef CAS PubMed.
- M. Armand, S. Grugeon, H. Vezin, S. Laruelle, P. Ribiere, P. Poizot and J. M. Tarascon, Nat. Mater., 2009, 8, 120 CrossRef CAS PubMed.
- T. Suga, S. Sugita, H. Ohshiro, K. Oyaizu and H. Nishide, Adv. Mater., 2011, 23, 751 CrossRef CAS PubMed.
- T. Suga, Y.-J. Pu, S. Kasatori and H. Nishide, Macromolecules, 2007, 40, 3167 CrossRef CAS.
- Y. Morishima, I. Akihara, H. S. Lim and S.-I. Nozakura, Macromolecules, 1987, 20, 978 CrossRef CAS.
- D. Clarke, B. C. Gilbert, P. Hanson and C. M. Kirk, J. Chem. Soc., Perkin Trans. 2, 1978, 1103 RSC.
- K. Oyaizu, Y. Ando, H. Konishi and H. Nishide, J. Am. Chem. Soc., 2008, 130, 14459 CrossRef CAS PubMed.
- (a) H. J. Dahms, J. Phys. Chem., 1968, 72, 362 CrossRef CAS; (b) I. Ruff and V. J. J. Friedrich, J. Phys. Chem., 1971, 75, 3297 CrossRef CAS.
- D. Sun, S. V. Rosokha and J. K. Kochi, J. Am. Chem. Soc., 2004, 126, 1388 CrossRef CAS PubMed.
- L. Levy, T. N. Tozer, L. D. Tuck and D. B. Loveland, J. Med. Chem., 1972, 15, 898 CrossRef CAS.
Footnote |
| † Electronic supplementary information (ESI) available: Details of the synthetic preparation and electrochemical characterization. See DOI: 10.1039/c4ra17107a |
| This journal is © The Royal Society of Chemistry 2015 |