
New insights into fluorinated TiO2 (brookite, anatase and rutile) nanoparticles as efficient photocatalytic redox catalysts†
Abstract
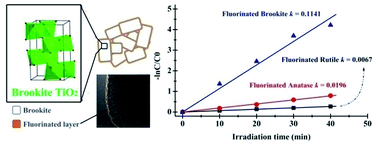
The synthesis of anionic-modified nanostructures with specific properties is often hindered by difficulty in tuning the material compositions without sacrificing phase purity and sample uniformity. Here, we present a novel methodology using NH4F as fluorine source and a high-temperature ionic diffusion that facilitate surface fluorination of TiO2 without changes of phase structure. Using this method, we prepared fluorinated TiO2 nanostructures of different phases (brookite, anatase and rutile) and investigated their structural and photocatalytic redox properties. Photocatalytic selective oxidation of MO and selective reduction of water in producing hydrogen under UV-light irradiation are employed as probe reactions to test the redox properties of the fluorinated TiO2 of different phases. The results show that the photocatalytic redox properties of TiO2 are highly dependent on phase structure and surface fluorination. Among all the phases, the oxidation activity of fluorinated TiO2 can be ranked as brookite > anatase > rutile. Strikingly, the photocatalytic reduction activity for these fluorinated TiO2 followed the same sequence. The variations of photocatalytic redox activity are most likely due to the synergism of phase composition control and surface fluorination. This could effectively delay electron–hole recombination, and generate more free ˙OH radicals but less free electrons. The methodology reported here is simple and general, which might be used to design and control the structures and properties of anionic-modified nanostructures for a variety of applications in environmental cleaning and energy conversion.
 Please wait while we load your content...
Please wait while we load your content...

