
Microwave role in the thermally induced SRN1 reaction for α-arylation of ketones†
Abstract
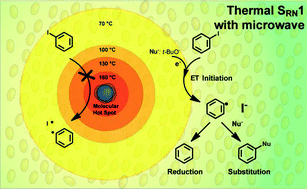
The coupling between iodobenzene and the enolate anion of acetophenone is accelerated by microwave irradiation. This increase in reaction rate is only ascribed to thermal effects. The coupling reaction gave the corresponding substitution product 1,2-di-phenylethanone in a 50% yield when microwave irradiation was applied between 15–60 s according to the intensity of the pulse. Moreover, this reaction is effective in a temperature window of 70–120 °C. The presence of ionic and dipolar species is not involved in the initiation process as molecular radiators. The excess of tBuOK in the reaction medium may also act as an electron donor helping to generate radicals when the solution temperature increases to 70 °C.
 Please wait while we load your content...
Please wait while we load your content...

