
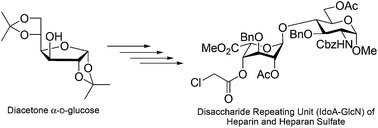
Formal synthesis of a disaccharide repeating unit (IdoA–GlcN) of heparin and heparan sulfate†
Abstract
A concise route to access the key disaccharide repeating unit (IdoA–GlcN) of heparan sulfate is described. The synthesis was accomplished by commercially available diacetone α-D-glucose to functional group transformations, which led to the formation of a L-iduronate donor. This L-iduronate donor was subsequently coupled with a glucosyl acceptor to form the corresponding key disaccharide repeating unit (IdoA–GlcN) of heparan sulfate in good overall yield.
 Please wait while we load your content...
Please wait while we load your content...

