
Suppression of protein aggregation by gold nanoparticles: a new way to store and transport proteins†
Anindita Dasa,
Abhijit Chakrabartib and
Puspendu K. Das*a
aDepartment of Inorganic and Physical Chemistry, Indian Institute of Science, Bangalore 560012, India. E-mail: pkdas@ipc.iisc.ernet.in; Fax: +91 8023600683; Tel: +91 8022932662
bCrystallography & Molecular Biology Division, Saha Institute of Nuclear Physics, 1/AF Bidhannagar, Kolkata 700064, India
First published on 20th April 2015
Abstract
Suppression of the aggregation of proteins has tremendous implications in biology and medicine. In the pharmaceuticals industry, aggregation of therapeutically important proteins and peptides while stored, reduces the efficacy and promptness of action leading to, in many instances, intoxication of the patient by the aggregate. Here we report the effect of gold nanoparticles (Au-NPs) in preventing the thermal and chemical aggregation of two unrelated proteins of different size, alcohol dehydrogenase (ADH, 84 kDa) and insulin (6 kDa), respectively, in physiological pH. Our principal observation is that there is a significant reduction (up to 95%) in the extent of aggregation of ADH and insulin in the presence of gold nanoparticles (Au-NPs). Aggregation of these proteins at micromolar concentration is prevented using nanomolar or less amounts of gold nanoparticles which is remarkable since chaperones which prevent such aggregation in vivo are required in micromolar quantity. The prevention of aggregation of these two different proteins under two different denaturing environments has established the role of Au-NPs as a protein aggregation prevention agent. The extent of prevention increases rapidly with the increase in the size of the gold nanoparticles. Protein molecules get physisorbed on the gold nanoparticle surface and thus become inaccessible by the denaturing agent in solution. This adsorption of proteins on Au-NPs has been established by a variety of techniques and assays.
Introduction
Aggregation is a critical issue for manufacturing, storage and transport of proteins and peptide based therapeutics. Aggregation includes formation of dimers and multimers in solution mediated by “apparently benign” solution conditions such as pH, salt concentration, temperature, and addition of preservatives.1 During preparation/isolation, shipping, storage and administration, protein and peptide based therapeutics undergo various chemical and physical changes that can adversely affect their functions and efficacy. Deamidation,2 denaturation, and oxidation which could lead to aggregation are some of the issues that were addressed on earlier studies regarding protein and peptide based therapeutics. Aggregation has emerged as a key issue having deleterious effects on protein and peptide based drugs leading to loss of potency, changed kinetics of release, reduced stability and thus, shelf-life.3–5Preventing or suppressing aggregation has thus become a major issue since current practice of pharmaceutical formulation toward high concentration protein solutions increases the protein–protein interaction leading to aggregation. Protein aggregation can increase the immunogenicity of a protein therapeutic and in some cases it can be life-threatening. For example, development of antibodies to erythropoietin generated by recombinant technology has been shown to produce “pure red cell aplasia” a life-threatening side – effect of the erythropoietin therapy in a number of patients receiving the recombinant product.6 Antibody response to aggregated human interferon alpha2b in wild type and transgenic mice depends on the type and extent of aggregation.7 Insulin is known to lose efficacy as a result of protein aggregation under high pressure or at low pH above room temperature.8,9 Due to injected aggregated insulin, patients develop antibodies to aggregated insulin but the antibodies turn out to be completely benign. As an important class of protein therapeutics, monoclonal antibodies in human clinical trials are on the rise. Patients often require high dosing as a convenient and comfortable mode of delivery. This needs high concentration of lyophilized monoclonal antibody, which makes it prone to aggregation. Lyophilization is very effective in stabilizing proteins.6 While oxidation, denaturation, and other types of instability may be prevented by lyophilization, aggregation and subsequent precipitation from solution still remains a major problem.9–11
The necessity of prevention of aggregation has led to the development of a number of excipients to reduce or eliminate aggregation and increase the solution stability of high-value protein therapeutics. One way to address the aggregation problem would be to add prevention agent in a concentrated solution of protein. But all such agents tried in the past will either cause toxicity or patient discomfort.12 While these excipients have been promoted by the drug companies in many protein and peptide based therapeutic formulations, they are difficult to synthesize in large quantities and are expensive. Cellular molecular chaperones, stress-induced proteins, and newly found chemical and pharmacological chaperones are also known to be effective for suppressing protein aggregation in micromolar concentrations.13–17 However, chaperones are difficult to synthesize, hard to purify, specific for each protein and costly. Therefore, an alternate and easy method is urgently needed.
A strategy for stopping protein aggregation could consist of a surface where a protein is adsorbed or collected, stored in the adsorbed state and released using an appropriate detachment mechanism from the surface when necessary. Interaction of micron size polystyrene latex particles with macromolecules was studied as early as 1966 by Wilkins and Myers and they found that surface modified latex particles have different mobility.18 Injecting colloids of different mobility into rats, they could control both the rate of clearance as well as the site of accumulation. Adsorption of selected macromolecules or proteins on colloidal gold was first reported by Geoghegan et al. in the late 1970s while trying to detect the presence of several sugar binding proteins on cell surface using electron microscopy.19 In their experiment the protein horseradish peroxidase (HRP) labeled with gold particles were added to peritoneal macrophages labeled with sugar binding protein Con-A. The location of HRP–Con A complex on the cell surface was found from the TEM images of the gold particles which were easily seen. Since these early studies, many reports on nanoparticle–protein interaction have appeared in the literature.20–22 The nanomaterial–protein/enzyme conjugates has been used in a diverse application ranging from decontamination to biosensing.23,24 Three critical factors that speak for the use of gold nanoparticles rather than other nanomaterials in medicine and diagnostics are its biocompatibility, lack of toxicity and ability to penetrate cells.25–30 Gold nanoparticles (Au-NPs) have been used extensively in cell modulation, drug delivery, diagnostic in cancer cell imaging and subsequent photothermal therapy,25,31–34 etc. Au-NPs provide large surface areas for facile association with a large range of molecules of biological interest such as amino acids,35 DNA,36 and proteins/enzymes.37 When proteins in a body fluid come in contact with Au-NPs, protein adsorption is the first step in a cascade of events that follow. Protein adsorption on Au-NPs and their subsequent conformation and activity changes have been studied extensively in the recent past.35,37,38 In 2008 De et al., have exploited the ability of the nanoparticle–protein interaction to refold the thermally denatured proteins. They have reported gold nanoparticle mediated regaining of the native conformation and thus activity of thermally denatured α-chymotrypsin, lysozyme and papain followed by their release from the nanoparticle surface by increasing the ionic strength.39 This report opened up the possibility of exploiting the role of nanoparticles as synthetic chaperones. During self-aggregation, proteins undergo unfolding which exposes the hydrophobic core.40 Conventional molecular chaperones interact with a non-native protein through their hydrophobic binding sites and assist it to refold.41 There are recent reports on resistance of protein, adsorbed on the surface of nanoparticles, towards aggregation in nonoptimal conditions.42,43 As the size of nanoparticles is known to be crucial for their interaction with biomolecules,37 the size must play an important role in prevention of protein aggregation on the surface of the Au-NPs in a denaturing environment. We have selected two unrelated test proteins, ADH and insulin which were led to undergo aggregation in vitro by two different denaturing methods, thermal and chemical respectively, in the presence and absence of Au-NPs. The formation of protein–nanoparticle conjugate and the prevention of protein aggregation have been characterized by a variety of scattering and spectroscopic assays and techniques.
Results and discussion
Effect of Au-NPs on thermal aggregation of ADH
Upon heating above 37 °C hydrophobic interaction which holds ADH in its native conformation breaks down leading to unfolding followed by aggregation.44 The conversion of ADH from its dispersed state to its aggregated form in solution can be monitored as a function of time by recording the scattered Rayleigh light intensity at 360 nm where native ADH is transparent. The time period for aggregation includes an initial lag phase for the nucleation of the native form when the overall scattered light intensity doesn't change upon heating. After nucleation, the ADH monomers undergo rapid association resulting in a near exponential growth in the size of the aggregates and the solution becomes turbid. During this association phase the scattered light intensity at 360 nm increases rapidly. Ultimately the scattered light intensity reaches saturation when aggregation is complete.45 To check the effect of Au-NPs on aggregation of ADH, Au-NPs were added to ADH solution prior to heating at 50 °C (Fig. 1) and the intensity of the scattered light was monitored. With addition of Au-NPs the scattering light intensity was much lower compared to pure ADH heated at 50 °C (Fig. 1) indicating lesser amount of aggregate formation in solution. The intensity of scattered light at saturation, when no Au-NPs were added to the ADH solution while it was heated at 50 °C, was taken as 100% aggregation and all scattering data were normalized with respect to that intensity. The data in Fig. 1 shows that 15 nm Au-NPs are able to prevent the thermal aggregation of ADH. When similar experiments were carried out with 30 nm Au-NPs keeping the total surface area same as those of 15 nm particles, scattered light intensity at saturation went down further. Then we carried out the experiments with larger size particles of 45 and 60 nm dia. keeping the total surface area the same. The quantitative result for prevention of aggregation and corresponding amounts of ADH and Au-NPs derived from Fig. 1 are displayed in Table 1. | ||
| Fig. 1 Aggregation of 0.23 mg mL−1 ADH upon heating at 50 °C in a final volume of 0.5 mL in the absence and presence of Au-NPs in 10 mM phosphate buffer (pH 7.0). Aggregation was monitored by time-dependent change in the scattering intensity at 360 nm. The curves denote ADH (a) alone and in the presence of (b) 15 nm (c) 30 nm (d) 45 nm and (e) 60 nm Au-NPs. | ||
| Conc. of ADH (mg mL−1) | Size of Au-NP (nm) | Conc. of Au-NP (nM) | Aggregation prevention (%) |
|---|---|---|---|
| 0.23 | 15 | 0.5 | 23 |
| 30 | 0.16 | 32 | |
| 45 | 0.065 | 49 | |
| 60 | 0.04 | 91 |
In order to show the lowering of scattered light intensity at 360 nm is not due to coagulation of Au-NPs in the presence of ADH several control experiments were carried out. The dispersed state of Au-NPs in water was verified by transmission electron microscope (TEM) image and UV-Vis spectra (Fig. S1 and S2 in ESI†). The state of aggregation of Au-NPs in buffer solutions of different composition was monitored by the surface plasmon resonance (SPR) peak position and broadening of the SPR spectra of Au-NPs in conjunction with the TEM images (Fig. S3 and S4 in ESI†). The addition of ADH in the solution of Au-NPs didn't change the absorption characteristics significantly (Fig. S5 in ESI†) indicating that the Au-NPs remain dispersed in buffer solution after protein addition. In a similar fashion the dispersion state of Au-NPs and Au-NP–ADH complex after heating to 50 °C has been verified (Fig. S6 and S7 in the ESI†). From all these control experiments it can be safely concluded that the decrease in the scattering intensity at 360 nm from the solution containing ADH and Au-NPs after heating to 50 °C in Fig. 1 is due to the suppression of thermal aggregation of ADH in the presence of Au-NPs.
There is a rapid nonlinear increase in the extent of prevention with increase in the size of the nanoparticles as shown in Fig. 2. The nonlinear increase in Fig. 2 is perhaps due to a progressive increase in the ease of binding of the protein molecules on a more flat surface. On a flat surface proteins can close-pack once it anchors on the binding site on the surface. In the best scenario they can all be aligned on the surface similar to a LB film. On a curved surface, the tails of the protein will be further apart and as a result the excluded volume after anchoring will be large. Consequently, in the vicinity of the surface the number of proteins that can be packed in a given volume will be lower on a curved surface than on a flat surface.
 | ||
| Fig. 2 Suppression of thermal aggregation of ADH as a function of size of the Au-NPs with the same total surface area in a final volume of 0.5 mL in 10 mM phosphate buffer (pH 7.0) at 50 °C. Aggregation was monitored by time-dependent change in the scattering intensity at 360 nm. The solid line leads the eye through the experimental points. (The horizontal and vertical bars indicate the error limits.) | ||
Clark and Huang while investigating the thermal aggregation of the eye lens protein, βL-crystallin at 60 °C have reported that the protein precipitates out from solution after reaching the saturation point causing a decrease in the scattered light intensity.44 Similar observation was made for the thermal denaturation of pure ADH at 56 °C by Markossian et al.46 The same thing happened in the scattered light signal observed during thermal denaturation of ADH in the absence and presence of Au-NPs as shown in Fig. 1. The onsets of aggregation and precipitation have different kinetics. These are reflected in the slopes of the curves in Fig. 1. The initiation of this precipitation depends on the size and concentration of the nanoparticles as well.
To further investigate the interaction of Au-NP surface with ADH we have done steady state fluorescence and dynamic light scattering (DLS) experiments. Fluorescence spectroscopy and imaging is a powerful technique for detection of tumor cells in vivo using an external fluorophore such as a dye molecule tagged with a target specific molecule.47,48 Quenching of the intrinsic fluorescence from tryptophan, tyrosine, phenylalanine residues by Au-NPs has also been used to investigate the binding of proteins on nanometer size surfaces using nanomaterial surface energy transfer (NSET).37,49
The fluorescence signal from ADH solution after excitation in the ultraviolet region in the absence and presence of 60 nm Au-NPs is displayed in Fig. 3. The tyrosine and tryptophan residues of ADH solution were excited at 280 nm50 and the subsequent emission at 336 nm was collected. The fluorescence intensity at 336 nm is found to decrease in the presence of nanoparticles, which is indicative of interaction between ADH and Au-NPs. But the position of the fluorescence band did not change pointing out that the conformation of ADH was not perturbed in the presence of Au-NPs. This quenching is widely known as the NSET in the literature. In NSET the energy transfer occurs from the molecular dipole of the fluorophore (here protein) to the surface of the nanomaterial.51 The extent of quenching increases with increasing concentration of Au-NPs. This quenching provides a strong evidence for the interaction of ADH with Au-NPs within a distance ≤40 nm which in the present case is achieved by the adsorption of ADH molecules onto the surface of Au-NPs.
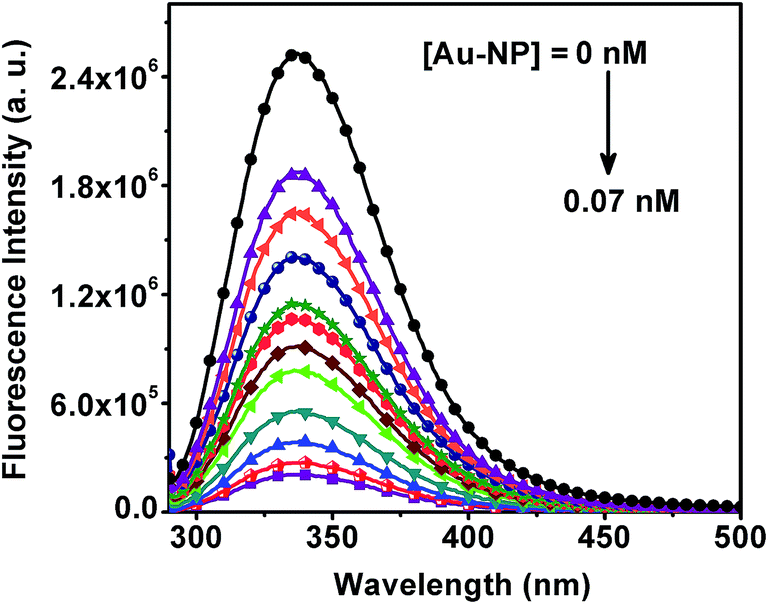 | ||
| Fig. 3 Quenching of fluorescence of ADH by Au-NPs. 0.23 mg mL−1 ADH in 10 mM pH 7.0 phosphate buffer were excited at 280 nm at 25 °C in the absence and presence of 60 nm Au-NP of different concentration. | ||
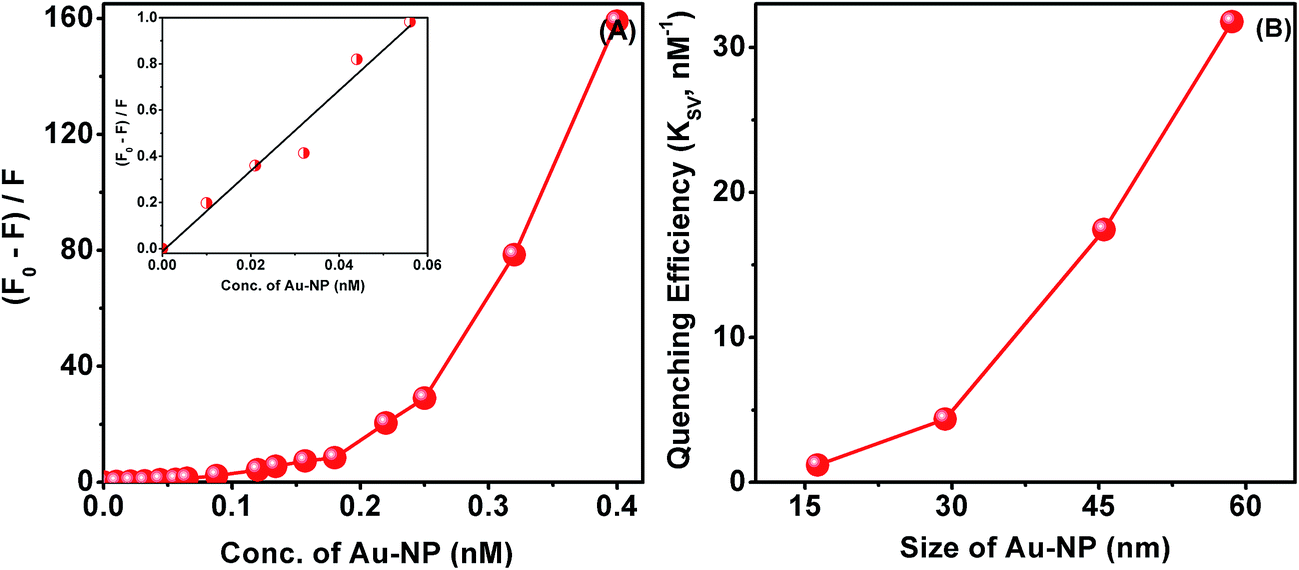
The efficiency of fluorescence quenching, which is a measure of the strength of the interaction of the protein with the nanoparticle surface, is dependent on the size of the Au-NPs. From the linear fitting of (F0 − F)/F on concentration of Au-NPs (Fig. 4A) we have estimated the relative kinetic efficiency of quenching, Ksv, based on the Stern–Volmer equation (see Experimental section). We have observed a nonlinear increase in Ksv with the size of Au-NPs, which is evident from Fig. 4B. This nonlinear variation of Ksv with size is consistent with the nonlinear size dependence of the extent of lowering of scattering intensity on saturation of aggregation. These data from scattering and fluorescence experiments lead to the only possible inference that the Au-NPs interact with the protein and the nature of the surface of the particle which varies with its size is involved in the interaction process. Since larger particles have flatter-surfaces, the protein which is in this case ADH assimilates better on a flat surface.
 | ||
| Fig. 4 (A) Relative quenching of fluorescence of 0.23 mg mL−1 ADH by 60 nm Au-NP (linear region of relative quenching of ADH fluorescence at lower concentration of nanoparticles is shown in inset). (B) Variation of Ksv with size. | ||
The association constant Ka, obtained from the Hill plot (described in the Experimental section) is seen to increase nonlinearly with the size of Au-NPs in Fig. 5A which indicates a higher extent of adsorption of ADH on the surface of bigger size nanoparticle. The degree of cooperativity, n, for all the four sizes of Au-NPs is close to one (Fig. 5B) which indicates that there is nearly one type of identical binding sites for the ADH on the Au-NP surface.
 | ||
| Fig. 5 (A) Variation of association constant and (B) degree of ADH–Au-NPs binding cooperativity (Hill coefficient) as a function of size of Au-NPs in 10 mM pH 7 phosphate buffer. | ||
The formation of protein–nanoparticle conjugate was further probed by DLS experiments where the hydrodynamic diameter DH of the nanoparticle was found to increase after addition of ADH. The results are summarized in Table 2.
| Au-NP (nm) | Hydrodynamic diameter of Au-NP (nm) | Hydrodynamic diameter of Au-NP + ADH (nm) | Increase in hydrodynamic diameter after ADH adsorption (nm) | Volume of the shell of adsorbed ADH molecules (nm3) |
|---|---|---|---|---|
| 30 | 27.9 | 49.0 | 21.1 | 50 229 |
| 45 | 47.1 | 63.8 | 16.7 | 81 266 |
| 60 | 61.6 | 72.0 | 10.4 | 73 044 |
From Table 2 it is seen that the DH of the Au-NPs increases after interaction with the protein and the increment in size is attributed to the thickness of the protein layer adsorbed on the Au-NP surface. The values of DH of Au-NPs and Au-NP–protein conjugates are qualitative since it depends on multiple variables such as pH, ionic strength of the medium, nature of the protein, surface property and the packing density of proteins on nanoparticles. In the context of our experiment where the medium, the protein and nanoparticles are fixed, the intensity of the scattered light intensity which determines the DH depends on the size and concentration of the nanoparticle. For a particular size there is a critical concentration below which the scattered light intensity is not sufficient to give a good distribution of hydrodynamic diameters. This critical concentration decreases with the size of nanoparticle.52 The hydrodynamic diameters of 15 nm Au-NPs before and after interaction with ADH were not shown due to poor signal quality under experimental concentrations that were employed in our prevention study. In Table 2 the volume of the shell of the adsorbed ADH molecules around the Au-NP is shown which has been calculated from the thickness of the adsorbed protein layer obtained from the DLS experiments. The volume is proportional to the number of ADH molecules adsorbed and depends on factors such as the density or packing of the protein layer, degree of hydration, etc. The change in volume was found to increase with size of Au-NPs. This observation justifies the increase in the extent of adsorption of ADH molecules on Au-NPs of bigger size responsible for the prevention of thermal aggregation of ADH to a large extent.
We have investigated the effect of concentration of Au-NPs on thermal aggregation of ADH at pH 7.0 at 50 °C and the results are graphically presented in Fig. 6. The quantitative result is presented graphically in Fig. 7.
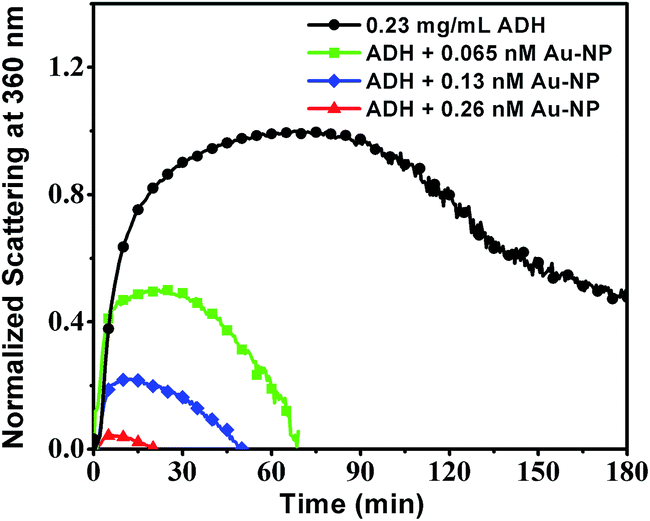 | ||
| Fig. 6 Aggregation of 0.23 mg mL−1 ADH which was initiated by heating at 50 °C in a final volume of 0.5 mL in the absence and presence of 45 nm Au-NPs in 10 mM phosphate buffer (pH 7.0) monitored by the time-dependent change in scattering at 360 nm. | ||
 | ||
| Fig. 7 Effect of concentration of 45 nm Au-NPs on ADH aggregation. Aggregation was initiated by heating at 50 °C in 0.5 mL solution of 0.23 mg mL−1 ADH in 10 mM phosphate buffer of pH = 7.0 (the results on extent of prevention are within ±5%, the error bar for the sizes are given in Fig. S1 in the ESI†). | ||
From Fig. 6 it is seen that the extent of prevention increases rapidly with concentration of the Au-NPs, which is expected since the total surface area for adsorption is larger at higher nanoparticle concentration. Therefore, the size and the concentration of the nanoparticles for a fixed concentration of the protein dictate the extent of prevention. For larger size particles one can achieve the same prevention efficiency at a lower concentration.
Effect of Au-NPs on dithiothreitol induced aggregation of insulin
In a similar set of experiments the effect of Au-NPs on dithiothreitol (DTT) induced aggregation of insulin was studied. Insulin is a smaller protein (∼6 kDa) compared to ADH (∼84 kDa) and is unrelated. Upon addition of DTT the disulfide bonds between A and B-chains of insulin undergo reduction followed by precipitation of insulin.16,41,53 The precipitation is due to the aggregation of the insulin B-chain. Hence the mechanisms for the thermal and DTT induced aggregation of proteins are entirely different. So the verification of the efficiency of Au-NPs to protect proteins under these two different denaturing conditions will establish the role of Au-NPs as protein aggregation suppressor, in general. With this aim the DTT induced aggregation of insulin has been studied in presence of 15–60 nm Au-NPs. The result of the experiments on prevention of aggregation of insulin by Au-NPs is displayed in Fig. 8. | ||
| Fig. 8 Aggregation of insulin upon addition of DTT in a final volume of 1 mL in the absence and presence of Au-NPs in 10 mM phosphate buffer (pH 7.0) at 25 °C. Aggregation was monitored by time-dependent change in the scattering intensity at 360 nm. The curves denote insulin (a) alone and in the presence of (b) 15 nm (c) 30 nm (d) 45 nm and (e) 60 nm Au-NPs. | ||
In literature plenty of reports are present to illustrate the ability of DTT to replace biomolecules like ss-DNA from the Au-NPs surface.54 On the contrary the cysteine rich protein like BSA adsorbed onto the Au-NPs surface is not displaced by DTT molecules present in solution.55 It is not known whether DTT can replace insulin adsorbed on the AuNP surface but there is a possibility that DTT binds with the nanoparticle surface and induce aggregation of nanoparticles in solution leading to a change in scattered light intensity at 360 nm. To demonstrate that the Au-NPs do not aggregate by addition of insulin and/or DTT, the UV-Visible spectra were recorded. The concentrations of Au-NPs were taken as the same as used in the prevention experiments. The results are shown in Fig. S8–S12 in the ESI.† In the resultant absorption spectra the SPR peak position and the width remained unchanged, which confirms that Au-NPs do not aggregate under these conditions. The result for prevention of aggregation and corresponding amounts of insulin and Au-NPs derived from Fig. 8 are displayed in Table 3.
| Conc. of insulin (mg mL−1) | Size of Au-NP (nm) | Conc. of Au-NP (nM) | Aggregation prevention (%) |
|---|---|---|---|
| 0.3 | 15 | 1 | 25 |
| 30 | 0.32 | 30 | |
| 45 | 0.13 | 50 | |
| 60 | 0.08 | 85 |
From Fig. 9 it is observed that the effect of size of Au-NPs on suppression of DTT induced aggregation of insulin follows a similar nonlinear behavior that we have seen in the case of ADH.
 | ||
| Fig. 9 Aggregation of insulin in the presence of 15–60 nm Au-NPs with constant total surface area in a final volume of 1 mL in 10 mM phosphate buffer (pH 7.0) at 25 °C. Aggregation was monitored by time-dependent change in the scattering intensity at 360 nm. The solid line connects the experimental data points (the horizontal and vertical bars indicate the error range. | ||
To further probe the nonlinear size dependence of aggregation, the fluorescence quenching studies were carried out for insulin in a manner similar to what was done for ADH. The fluorescence signal from insulin solution after excitation in the ultraviolet region, which originates from four tyrosine residues16 in the absence and presence of 45 nm Au-NPs has been displayed in Fig. 10. Insulin was excited at 280 nm and the subsequent emission at 305 nm was collected. The emission maximum at 305 nm did not shift significantly with addition of Au-NPs, but the intensity decreased. The decrease in intensity indicates that the Au-NP is interacting with insulin. However, the interaction did not affect the native conformation of insulin thereby keeping the position of the emission maximum unchanged.
 | ||
| Fig. 10 Quenching of fluorescence of insulin by Au-NPs. Tyrosine residues of 0.3 mg mL−1 insulin in 10 mM pH 7.0 phosphate buffer were excited at 280 nm at 25 °C in the absence and presence of 45 nm Au-NPs. | ||
A non-linear increase of Ksv with the size of the Au-NPs, as seen in the case of ADH, is observed (Fig. 11) for insulin although the extent of increase is different for these two proteins. This is expected since insulin being a small protein interact differently with nanoparticles from ADH which is a moderate size protein.
 | ||
| Fig. 11 (A) Relative quenching of fluorescence of 0.3 mg mL−1 insulin by 45 nm Au-NPs (linear region of relative quenching of insulin fluorescence at lower concentration of nanoparticles has been shown in the inset). (B) Variation of Ksv with the size of Au-NPs. | ||
The same trends of nonlinear increase of Ka and n have been observed for insulin interaction with Au-NPs of different sizes in Fig. 12 as seen in the case of ADH interaction with Au-NPs. This nonlinear increase in the association constant with size of Au-NPs establish the general role of size of Au-NPs on the extent of adsorption of proteins which in turn is related to their protein aggregation prevention capacity. The value of Hill coefficient for the interaction of insulin with Au-NPs for all the four different sizes indicates the near identical nature of binding sites of insulin on the surface of Au-NPs.
 | ||
| Fig. 12 Aggregation of insulin in the absence and presence of different concentrations of 45 nm spherical Au-NPs. Aggregation of insulin in 10 mM phosphate buffer (pH 7.0) at 25 °C was initiated by addition of DTT in a final volume of 1 mL in the absence and presence of Au-NPs. The appearance of turbidity was monitored by the time-dependent change in scattering intensity at 360 nm. | ||
The effect of concentration of 45 nm spherical Au-NPs on DTT induced aggregation of insulin is graphically shown in Fig. 13.
 | ||
| Fig. 13 Aggregation of insulin in the absence and presence of different concentrations of 45 nm spherical Au-NPs. Aggregation of insulin in 10 mM phosphate buffer (pH 7.0) at 25 °C was initiated by addition of DTT in a final volume of 1 mL in the absence and presence of Au-NPs. The appearance of turbidity was monitored by the time-dependent change in scattering intensity at 360 nm. | ||
Surface charge of nanoparticle–protein conjugate
The interaction of Au-NPs with proteins was further probed by measuring the zeta potential of Au-NPs before and after their interaction. The zeta potential of the nanoparticles indicates the charge and its polarity on the surface. Since the nanoparticles are negatively charged, the zeta potential is negative. The results are summarized in Table 4. The zeta potential depends on the size and concentration of the Au-NPs,56,57 pH,58 and ionic strength of the medium,59 the nature of the capping agent on Au-NPs,60 and the characteristics of the protein in solution.61| Protein | Conc. of protein (mg mL−1) | Zeta potential of protein (mV) | Size of Au-NP (nm) | Conc. of Au-NP (nM) | Zeta potential of Au-NP (mV) | Zeta potential of protein + Au-NP (mV) |
|---|---|---|---|---|---|---|
| ADH | 0.23 | −2.4 | 15 | 0.5 | −16.2 | −7.0 |
| 30 | 0.16 | −15.2 | −6.4 | |||
| 45 | 0.065 | −39.1 | −7.7 | |||
| 60 | 0.04 | −28.2 | −7.6 | |||
| Insulin | 0.3 | −7.2 | 15 | 1 | −34.8 | −28.7 |
| 30 | 0.32 | −21.3 | −16.6 | |||
| 45 | 0.13 | −41.6 | −37.4 | |||
| 60 | 0.08 | −38.9 | −27.5 |
From Table 4 we see that the zeta potential of ADH is less negative than that of insulin at the experimental pH 7.0 since insulin exists in its anionic form while ADH is not fully ionized at this pH. The isoelectric point, pI of ADH is 6.8 while that of insulin is 5.3.62,63 Thus at pH 7 the proteins with lower pI will be ionized to a greater extent. The zeta potential of Au-NPs becomes less negative after interaction with cysteine rich insulin and ADH since the protein adsorption on the nanoparticle surface occurs via replacement of the citrate ions by cysteines.64,65 The difference of zeta potentials of the same size particles in the two set of experiments is due to the difference in concentrations of the Au-NPs used in the experiments.
Effect of Au-NPs on the secondary structure of the protein
It is important to know the secondary structure of the protein in solution before and after interaction with Au-NPs and investigate the effect of Au-NPs on the secondary structure of the protein. The secondary structure of the protein can be obtained from far-UV CD spectrum as described in the literature.66The secondary structure of the enzyme ADH with a typical CD spectrum of a α/β-protein67,68 is retained below 60 °C as reported earlier. The far-UV CD spectra of ADH in the presence of Au-NPs at room temperature and after incubation for 2 h at 50 °C are shown in Fig. 14A and B, respectively. It is apparent that Au-NPs do not change the secondary structure of ADH significantly.
 | ||
| Fig. 14 Far-UV CD spectra of 0.23 mg mL−1 ADH in 10 mM pH 7.0 phosphate buffer (A) at 25 °C in the absence and presence of 15–60 nm Au-NPs, (B) heating at 50 °C for 2 h in the absence and presence of 15 or 45 nm Au-NPs. | ||
From Fig. 15A it is seen that the secondary structure of insulin at room temperature is rich in α-helix, which is characterized by two minima at 222 nm and 208 nm and a maxima at 195 nm in the CD spectrum.67,69 The CD spectrum of insulin remains unaltered in the presence of Au-NPs of different size implying that the Au-NPs do not change the secondary structure of the protein. When DTT is added to the solution the CD spectrum of insulin changes drastically as seen from Fig. 15B. Addition of DTT leads to the breakdown of the helical structure of insulin leading to randomization of the structure. This is expected since DTT reduces the disulfide bonds in insulin which are exposed in the medium even when it is adsorbed onto the Au-NP surface resulting in the breakdown of its secondary structure.
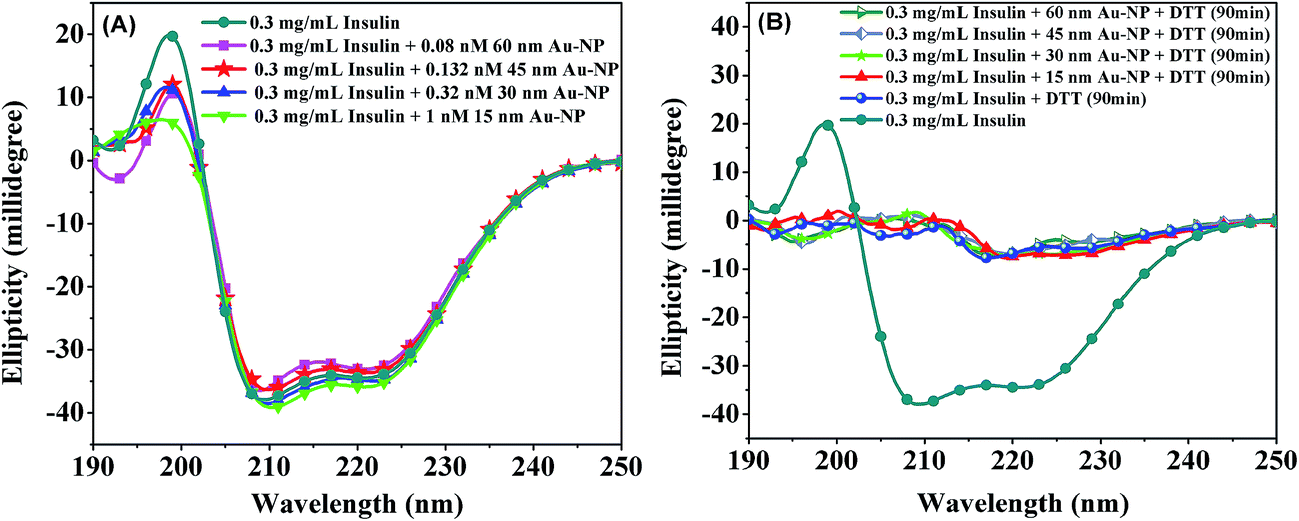 | ||
| Fig. 15 Far-UV CD spectra of 0.3 mg mL−1 insulin in 10 mM pH 7.0 phosphate buffer at 25 °C in the presence of Au-NPs (A), and after 90 min of addition of DTT (B). | ||
The fact that secondary structures of ADH and insulin remain unchanged in the adsorbed state is significant since there are many reports in the literature where it has been demonstrated that proteins when adsorbed on the nanoparticle surface undergo changes in their secondary structure.70,71 However, Fig. 15B shows that for adsorbed insulin on gold nanoparticles the secondary structure is no longer preserved when the chemical denaturant, DTT is added to the solution. In an earlier work Fischer et al. bound and denatured α-chymotrypsin by carboxylic acid functionalized anionic AuNPs and then released the protein from the surface by additing cationic surfactants such as derivatives of trimethylamine-functionalized surfactants.20 Based on their observations, De et al. have hypothesized that highly charged nanoparticle based hosts could serve as refolding agents by interacting with charged residues of denatured proteins accelerating refolding.39 Although we could not ascertain if the adsorbed insulin which were denatured upon DTT treatment could refold when detached from the nanoparticles, their observations39 in conjunction with our results on both ADH and insulin bring out the importance of using capped and functionalized Au-NPs as metallic chaperones in the context of refolding and protein aggregation prevention ability, in general.
Conclusion
In order to provide evidence that suppression of protein aggregation depends on the size, gold nanoparticles of 15–60 nm dia. have been used, in this paper, to investigate aggregation of two unrelated proteins, ADH and insulin in their presence. We have used a variety of scattering and spectroscopic assays in a multipronged approach to investigate the influence of size of gold nanoparticles on the aggregation of ADH and insulin induced by different methods. Rayleigh scattering, steady-state fluorescence, dynamic light scattering, and zeta potential measurements have provided clear evidence that gold nanoparticles prevent protein aggregation by adsorbing them on their surface. Adsorption makes the protein molecules inaccessible by denaturing agents/methods. We have shown that nanomolar or even sub-nanomolar concentration of Au-NPs could prevent aggregation of micromolar solutions of ADH or insulin under physiological conditions. The extent of prevention is significant and the effect increases with the size and concentration of the nanoparticles. Denaturation followed by aggregation is a serious problem in storage and transport of proteins and peptide therapeutics over a long period of time. In nature, chaperones prevent newly synthesized proteins from misfolding and/or aggregation and assist them to fold correctly inside cells. However, chaperones are very specific for specific proteins and typically micromolar quantities of chaperones are necessary for assisting proteins to fold correctly and prevent aggregation. We have demonstrated that at least the last function of chaperones, that is, aggregation prevention is served by gold nanoparticles and that too at much lower concentrations. This work opens up a new possibility for replacement of conventional chaperones which regulates protein aggregation at the cellular level by Au-NPs. In the light of growing importance of pharmaceutical use of proteins in biotechnology and medicine, Au-NPs could be very useful acting as metallic chaperones in improving solubilisation at high protein concentrations, the stability and shelf-life of purified and recombinant proteins.Other noble metal particles like silver, copper, etc. may exhibit a similar property which remains to be explored.
Experimental section
Materials
Insulin from porcine pancreas, ADH from equine liver, HAuCl4·3H2O and DTT were purchased from Sigma-Aldrich and were used without further purification. Ascorbic acid, tri-sodium citrate, NaOH, potassium dihydrogen phosphate and dipotassium hydrogen phosphate were obtained locally.Synthesis of Au-NPs
There are many methods for the synthesis of Au-NPs72–74 although control of shape during preparation of larger particles is still an unsolved issue. Some of the methods are very shape specific during formation of bigger structures and some involve time-consuming cleaning steps or toxic and hazardous side products to the environment.74,75 We have chosen the method described by Ziegler et al.,76 with modifications as appropriate. Spherical shape Au-NPs of diameters ranging from 15–60 nm were prepared by seed mediated synthesis using biocompatible reducing and capping agents.The synthesis of the nanoparticle in brief is as follows: 2.5 mL of HAuCl4·3H2O solution (0.2% w/v) in 50 mL of water was heated to boiling in a double necked round bottom flask and 2 mL of sodium-citrate solution (1% w/v, containing 0.05% w/v citric acid) was added quickly to the boiling solution under vigorous stirring. When the color of the solution turned from yellow to red, the solution was further kept boiling for 10 min and then allowed to cool down to room temperature. This resulted in 15 nm Au-NPs which we have used as seeds for the next step growth.
The general procedure followed for the preparation of large size nanoparticles using 15 nm seeds in first step growth is as follows. A certain amount of seed solution diluted to 20 mL was placed into a round bottom flask. To this a 10 mL aliquot of the precursor solution A containing HAuCl4·3H2O, and 10 mL of the solution B containing trisodium citrate as capping and ascorbic acid as reducing agent, were added at room temperature over an hour under vigorous stirring. After the addition was complete, the solution in the round bottom flask was heated to boiling. The solution was boiled for about 30 min and then allowed to cool down to room temperature. By this method we have prepared 30 and 45 nm Au-NPs using different volumes of HAuCl4·3H2O (0.2% w/v) in solution A which was determined from the density of gold fcc crystal, volume difference between required size and seed nanoparticles. The procedure for preparation of 60 nm size Au-NPs was carried out via second growth step where the 45 nm Au-NPs of the first step growth is used as seed solution. The 45 nm Au-NPs of first step growth was first diluted to 20 mL and then a 10 mL aliquot of the precursor solution A and 10 mL of the solution B as described above, were added slowly at room temperature over an hour under vigorous stirring. After the addition was complete, the solution was heated to boiling and maintained for 4 h. The solution was then allowed to cool down.
In solution A HAuCl4·3H2O stock solution (0.2% w/v) was diluted to 10 mL and in solution B an ascorbic acid stock solution (1% w/v) and a trisodium-citrate stock-solution (1% w/v) were mixed and diluted to 10 mL and the relative volume ratios of HAuCl4·3H2O, ascorbic acid and tri-sodium citrate stock solutions were always kept at 8![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) :2:1.
:2:1.
The prepared Au-NPs were characterized by transmission electron microscopy and UV-Vis spectroscopy. Electron microscope images of differently sized Au-NPs show their spherical geometry with very low spread in size (see Fig. S1 in ESI†). The UV-Vis spectra of the Au-NPs were shown in ESI (Fig. S2†). The concentration of the Au-NPs was estimated using the concentration of HAuCl4 used for the nanoparticle synthesis and diameter of the nanoparticle formed as described by Liu et al.77
Transmission electron microscopy of Au-NPs
Bright field transmission electron micrographs of freshly prepared Au-NPs were taken in JEOL 100CXII transmission electron microscope (TEM) with 100 kV accelerating voltage. Sample preparation was done by drop coating Au-NPs solution on formvar carbon film on 200 mesh copper grids (see Fig. S1 in ESI† for TEM images and histogram of size distribution of Au-NPs).Spectra of Au-NPs
Au-NPs of size 15–60 nm were characterized by a Perkin-Elmer Lambda-750 UV-Vis spectrophotometer by monitoring the surface plasmon resonance (SPR) peak position and width in the wavelength range 200–800 nm with a slit – width 2 nm (see Fig. S2 in ESI† for UV-Vis spectra of Au-NPs).The stability of metallic nanoparticles in phosphate buffer of pH 7.0 with different buffer concentrations was studied by monitoring the surface plasmon peak position and broadening of the UV-Vis spectroscopy (Fig. S3 in ESI†) and transmission electron microscopy (Fig. S4 ESI†).
Light scattering assay for aggregation
The scattered light intensity was monitored at 360 nm where protein solution is transparent. For ADH aggregation the scattered light intensity was monitored over 3 h in a Perkin-Elmer Lambda-750 UV-Vis spectrophotometer with a slit width of 2 nm.For the insulin aggregation the scattered light intensity was monitored over a time of 90 min in a Perkin-Elmer Lambda-35 UV-Vis spectrophotometer with a slit width 2 nm.
Thermal aggregation of ADH in the absence and presence of Au-NPs
A stock solution of ADH was prepared by dissolving ADH in 1 mL of 10 mM phosphate buffer of pH = 7.0. Then the stock was diluted to a concentration of 0.23 mg mL−1 in 0.5 mL of 10 mM phosphate buffer at pH 7.0 in the presence and absence of 15–60 nm sized citrate capped spherical Au-NPs. The aggregation of ADH was induced by heating the solution at 50 °C in the absence and presence of Au-NPs in a thermostat attached with the spectrophotometer and Julabo temperature controller. The aggregation of ADH was measured as a function of time at 50 °C by monitoring the scattering at 360 nm over a time of 3 h.Aggregation of insulin in the absence and presence of Au-NPs
A stock solution of insulin was prepared by dissolving insulin in a small volume of 20 mM NaOH followed by 10 mM phosphate buffer of pH = 7.0. The solution was centrifuged and the supernatant was collected to separate the insoluble part. This supernatant was then diluted to 1 mL to a concentration of 0.3 mg mL−1 in 10 mM phosphate buffer at pH 7.0 in the presence and absence of 15–60 nm citrate capped spherical Au-NPs. The reduction of insulin was initiated by adding 20 μL of 1 M Dithiothreitol (DTT) to 1 mL of (0.3 mg mL−1) insulin in the absence and presence of Au-NPs. The aggregation of insulin was measured as a function of time at 25 °C. The amount of DTT was sufficient for reduction of insulin even in the presence of nanoparticles in solution which was established with Fourier transform infrared spectroscopy (FTIR) of solution containing DTT in the absence and presence of Au-NPs (ESI Fig. S13†). The IR spectra were recorded in the transmission mode in a Vertex-70 FTIR spectrophotometer (Bruker Optik) with 0.5 cm−1 resolution and the signal was averaged over 512 scans. For recording the IR spectrum, a thin film of the sample on a CaF2 window was prepared by placing a few drops of the solution followed by drying. The window was placed in the cell holder of the spectrophotometer.As there is sufficient absorbance of Au-NPs at 360 nm, any aggregation of Au-NPs in the medium due to dispersion in phosphate buffer, interaction with ADH, insulin, or DTT may give rise to change in scattering at 360 nm. To insure the stability of nanoparticles during scattering experiments we have monitored the scattered light intensity at 360 nm for 1 h in control experiments such as a) nanoparticles in 10 mM pH = 7.0 phosphate buffer at 25 °C (data not shown), b) protein and nanoparticles in 10 mM pH = 7.0 phosphate buffer at 25 °C (Fig. S5 and S8 in ESI†), c) DTT and nanoparticles in 10 mM pH = 7.0 phosphate buffer at 25 °C (Fig. S9 and S12 in ESI†) and d) nanoparticles in 10 mM pH = 7.0 phosphate buffer at 50 °C (Fig. S6 in ESI†). It was observed that although there was no significant change in the scattered light intensity in the controls “a”, “b” and “c” mentioned above. There was a slight decrease in the intensity in control “d” due to a decrease in the surface Plasmon resonance absorption of Au-NPs as a function of increasing temperature.78 To nullify the effect of temperature the reference for studying ADH aggregation in the presence of Au-NPs at 50 °C was taken as nanoparticles in buffer at 50 °C.
Fluorescence quenching measurements
Intrinsic fluorescence of the tryptophan residues of alcohol dehydrogenase and tyrosine residues of insulin were monitored in the absence and presence of Au-NPs in 10 mM pH 7.0 phosphate buffer at 25 °C in Horiba Jobin Yvon Fluoromax-4 Spectrofluorometer. The fluorophores were excited at 280 nm and the emission was monitored in the range 290–500 nm. The excitation and emission band passes were kept at 5 nm. The concentration of alcohol dehydrogenase was kept at 0.23 mg mL−1 and the concentration of the insulin was kept at 0.3 mg mL−1. The concentration of Au-NPs was varied in the range 5–0.2 nM (15 nm Au-NPs), 0.6–0.05 nM (30 nm Au-NPs), 0.4–0.01 nM (45 nm Au-NPs), 0.07–0.005 nM (60 nm Au-NPs).The Stern–Volmer model which is applicable at lower concentration of the quencher,37 helps determine the Stern–Volmer constant (Ksv), i.e. relative kinetic efficiency of quenching. The Ksv is the slope of the plot of (F0 − F)/F vs. concentration of quencher (here, Au-NPs) where F0 and F are the fluorescence intensities of the protein in the absence and presence of quencher, respectively.
As multiple protein molecules interact with the Au-NPs in solution, the association constant (Ka) and degree of cooperativity (n) of the adsorption of protein on the nanoparticle surface can be represented by the Hill equation.
 | (1) |
 | (2) |
 | (3) |
Dynamic light scattering and zeta potential measurements
The Dynamic Light Scattering (DLS) and zeta potential measurements were performed in Malvern-Zetasizer Nano ZS90 apparatus. For DLS experiment 1 mL solution of Au-NPs was taken in a 3 mL transparent quartz cuvette and for the zeta potential measurement 1 mL solution of Au-NPs was taken in a disposable zeta cell. Data were recorded after attaining the thermal equilibrium at 25 °C. For DLS and zeta potential measurements with ADH the composition of nanoparticles and proteins were maintained at the same levels as given in Table 1. For zeta potential measurements with insulin the composition of nanoparticles and proteins were maintained at the same levels as given in Table 3. The DLS results for insulin Au-NPs has not been shown.Far-UV circular dichroism
The Far-UV circular dichroism (CD) measurements were performed in a JASCO J-715 spectropolarimeter. The solution of proteins in 10 mM pH 7 phosphate buffer in the absence and presence of 15–60 nm Au-NPs, with composition as given in Tables 1 and 3, were taken in a 350 μL volume and 1 mm pathlength quartz cuvette. The thermal denaturation of ADH was performed by heating the solution in an external heat bath at 50 °C and then monitoring the protein conformation after certain intervals in the CD instrument. The conformation of insulin was also monitored in a denaturing environment by addition of 20 μL 1 M DTT.Acknowledgements
P. K. Das thanks the Ministry of Communication and Information Technology for the Centre of Excellence in Nanoelectronics, Phase II grant for generous funding of this work. We thank S. Subramanian for letting us use the DLS instrument for carrying out our experiments.References
- E. Y. Chi, S. Krishnan, T. W. Randolph and J. F. Carpenter, Pharm. Res., 2003, 20, 1325–1336 CrossRef CAS.
- N. E. Robinson and A. B. Robinson, Proc. Natl. Acad. Sci. U. S. A., 2001, 98, 12409–12413 CrossRef CAS PubMed.
- G. Jiang, B. H. Woo, F. Kang, J. Singh and P. P. DeLuca, J. Controlled Release, 2002, 79, 137–145 CrossRef CAS PubMed.
- A. S. Rosenberg, AAPS J., 2006, 8, E501–E507 CrossRef PubMed.
- M. A. Alsenaidy, N. K. Jain, J. H. Kim, C. R. Middaugh and D. B. Volkin, Front. Pharmacol., 2014, 5, 39 Search PubMed.
- N. Casadevall, J. Nataf, B. Viron, A. Kolta, J.-J. Kiladjian, P. Martin-Duront, P. Michaud, T. Papo, V. Ugo, I. Teyssandier, B. Varet and P. Mayeux, N. Engl. J. Med., 2002, 346, 469–475 CrossRef CAS PubMed.
- S. Hermeling, H. Schellekens, C. Maas, M. F. B. G. Gebbink, D. J. A. Crommelin and W. Jiskoot, J. Pharm. Sci., 2006, 95, 1084–1096 CrossRef CAS PubMed.
- W. Dzwolak, R. Ravindra, J. Lendermann and R. Winter, Biochemistry, 2003, 42, 11347–11355 CrossRef CAS PubMed.
- J. Haas, E. Vöhringer-Martinez, A. Bögehold, D. Matthes, U. Hensen, A. Pelah, B. Abel and H. Grubmüller, ChemBioChem, 2009, 10, 1816–1822 CrossRef CAS PubMed.
- F. E. Dische, C. Wernstedt, G. T. Westermark, P. Westermark, M. B. Pepys, J. A. Rennie, S. G. Gilbey and P. J. Watkins, Diabetologia, 1988, 31, 158–161 CrossRef CAS PubMed.
- E. T. Maggio, BioProcess Int., 2008, 6, 58–65 CAS.
- J. C. S. Kim and L. S. Kaminsky, Toxicol. Pathol., 1988, 16, 35–45 CrossRef CAS PubMed.
- B. Raman, T. Ramakrishna and C. M. Rao, FEBS Lett., 1995, 365, 133–136 CrossRef CAS PubMed.
- S. Guha, T. K. Manna, K. P. Das and B. Bhattacharyya, J. Biol. Chem., 1998, 273, 30077–30080 CrossRef CAS PubMed.
- T. K. Chaudhuri and S. Paul, FEBS J., 2006, 273, 1331–1349 CrossRef CAS PubMed.
- N. L. Vekshin, Biochemistry, 2008, 73, 458–462 CAS.
- K. Giger, R. P. Vanam, E. Seyrek and P. L. Dubin, Biomacromolecules, 2008, 9, 2338–2344 CrossRef CAS PubMed.
- D. J. Wilkins and P. A. Myers, Br. J. Exp. Pathol., 1966, 47, 568–576 CAS.
- W. D. Geoghegan and G. A. Ackerman, J. Histochem. Cytochem., 1977, 25, 1187–1200 CrossRef CAS PubMed.
- N. O. Fischer, A. Verma, C. M. Goodman, J. M. Simard and V. M. Rotello, J. Am. Chem. Soc., 2003, 125, 13387–13391 CrossRef CAS PubMed.
- Z. G. Peng, K. Hidajat and M. S. Uddin, J. Colloid Interface Sci., 2004, 271, 277–283 CrossRef CAS PubMed.
- M.-E. Aubin-Tam and K. Hamad-schifferli, Langmuir, 2005, 21, 12080–12084 CrossRef CAS PubMed.
- A. S. Campbell, C. Dong, J. S. Dordick and C. Z. Dinu, Process Biochem., 2013, 48, 1355–1360 CrossRef CAS.
- A. S. Campbell, C. Dong, F. Meng, J. Hardinger, G. Perhinschi, N. Wu and C. Z. Dinu, ACS Appl. Mater. Interfaces, 2014, 6, 5393–5403 CAS.
- R. Shukla, V. Bansal, M. Chaudhary, A. Basu, R. R. Bhonde and M. Sastry, Langmuir, 2005, 21, 10644–10654 CrossRef CAS PubMed.
- J. A. Khan, B. Pillai, T. K. Das, Y. Singh and S. Maiti, ChemBioChem, 2007, 8, 1237–1240 CrossRef CAS PubMed.
- X. Zhang, W. Hu, J. Li, L. Tao and Y. Wei, Toxicol. Res., 2012, 1, 62–68 RSC.
- S. Vijayakumar and S. Ganesan, J. Nanomater., 2012, 2012, 734398 Search PubMed.
- R. D. Lima, A. B. Seabra and N. Duran, J. Appl. Toxicol., 2012, 32, 867–879 CrossRef CAS PubMed.
- C. E. Bradburne, J. B. Delehanty, K. B. Gemmill, B. C. Mei, H. Mattoussi, K. Susumu, J. B. Blanco-Canosa, P. E. Dawson and I. L. Medintz, Bioconjugate Chem., 2013, 24, 1570–1583 CrossRef CAS PubMed.
- C. Loo, A. Lowery, N. Halas, J. West and R. Drezek, Nano Lett., 2005, 5, 709–711 CrossRef CAS PubMed.
- M. Everts, V. Saini, J. L. Leddon, R. J. Kok, M. Stoff-khalili, M. A. Preuss, C. L. Millican, G. Perkins, J. M. Brown, H. Bagaria, D. E. Nikles, D. T. Johnson, V. P. Zharov and D. T. Curiel, Nano Lett., 2006, 6, 587–591 CrossRef CAS PubMed.
- D. R. Bhumkar, H. M. Joshi, M. Sastry and V. B. Pokharkar, Pharmaceut. Res., 2007, 24, 1415–1426 CrossRef CAS PubMed.
- S. Dhar, W. L. Daniel, D. A. Giljohann, C. A. Mirkin and S. J. Lippard, J. Am. Chem. Soc., 2009, 131, 14652–14653 CrossRef CAS PubMed.
- C. You, M. De, G. Han and V. M. Rotello, J. Am. Chem. Soc., 2005, 127, 12873–12881 CrossRef CAS PubMed.
- X. Zhang, B. Liu, M. R. Servos and J. Liu, Langmuir, 2013, 29, 6091–6098 CrossRef CAS PubMed.
- S. H. D. P. Lacerda, J. J. Park, C. Meuse, D. Pristinski, M. L. Becker, A. Karim and J. F. Douglas, ACS Nano, 2010, 4, 365–379 CrossRef PubMed.
- X. Jiang, J. Jiang, Y. Jin, E. Wang and S. Dong, Biomacromolecules, 2005, 6, 46–53 CrossRef CAS PubMed.
- M. De and V. M. Rotello, Chem. Commun., 2008, 3504–3506 RSC.
- J. I. Clark and P. J. Muchowski, Curr. Opin. Struct. Biol., 2000, 10, 52–59 CrossRef CAS PubMed.
- M. Bhattacharyya, S. Ray, S. Bhattacharya and A. Chakrabarti, J. Biol. Chem., 2004, 279, 55080–55088 CrossRef CAS PubMed.
- S. D. Luthuli, M. M. Chili, N. Revaprasadu and A. Shonhai, IUBMB Life, 2013, 65, 454–461 CrossRef CAS PubMed.
- V. Banerjee and K. P. Das, Langmuir, 2014, 30, 4775–4783 CrossRef CAS PubMed.
- J. I. Clark and Q.-L Huang, Proc. Natl. Acad. Sci. U. S. A., 1996, 93, 15185–15189 CrossRef CAS.
- S. Auer, C. M. Dobson and M. Vendruscolo, HFSP J., 2007, 1, 137–146 CrossRef CAS PubMed.
- K. A. Markossian, N. V. Golub, H. A. Khanova, D. I. Levitsky, N. B. Poliansky, K. O. Muranov and B. I. Kurganov, Biochim. Biophys. Acta, Gen. Subj., 2008, 1784, 1286–1293 CrossRef CAS PubMed.
- Z. Yu, R. M. Schmaltz, T. C. Bozeman, R. Paul, M. J. Rishel, K. S. Tsosie and S. M. Hecht, J. Am. Chem. Soc., 2013, 135, 2883–2886 CrossRef CAS PubMed.
- B. R. Schroeder, M. I. Ghare, C. Bhattacharya, R. Paul, Z. Yu, P. A. Zaleski, T. C. Bozeman, M. J. Rishel and S. M. Hecht, J. Am. Chem. Soc., 2014, 136, 13641–13656 CrossRef CAS PubMed.
- D. Gao, Y. Tian, S. Bi, Y. Chen, A. Yu and H. Zhang, Spectrochim. Acta, Part A, 2005, 62, 1203–1208 CrossRef PubMed.
- P. L. Luisi and R. Favilla, Eur. J. Biochem., 1970, 17, 91–94 CrossRef CAS PubMed.
- J. Griffin, A. K. Singh, D. Senapati, P. Rhodes, K. Mitchell, B. Robinson, E. Yu and P. C. Ray, Chem.–Eur. J., 2009, 15, 342–351 CrossRef CAS PubMed.
- J. Lim, S. P. Yeap, H. X. Che and S. C. Low, Nanoscale Res. Lett., 2013, 8, 381 CrossRef PubMed.
- J. M. Souza, B. I. Giasson, V. M.-Y. Lee and H. Ischiropoulos, FEBS Lett., 2000, 474, 116–119 CrossRef CAS PubMed.
- S. J. Hurst, A. K. R. Lytton-Jean and C. A. Mirkin, Anal. Chem., 2006, 78, 8313–8318 CrossRef CAS PubMed.
- D.-H. Tsai, M. P. Shelton, F. W. DelRio, S. Elzey, S. Guha, M. R. Zachariah and V. A. Hackley, Anal. Bioanal. Chem., 2012, 404, 3015–3023 CrossRef CAS PubMed.
- R. Tantra, P. Schulze and P. Quincey, Particuology, 2010, 8, 279–285 CrossRef CAS.
- S.-H. Wang, C.-W. Lee, A. Chiou and P.-K. Wei, J. Nanobiotechnol., 2010, 8, 33–46 CrossRef CAS PubMed.
- L. Sun, Z. Zhang, S. Wang, J. Zhang, H. Li, L. Ren, J. Weng and Q. Zhang, Nanoscale Res. Lett., 2009, 4, 216–220 CrossRef CAS PubMed.
- G. A. Rance and A. N. Khlobystov, Phys. Chem. Chem. Phys., 2010, 12, 10775–10780 RSC.
- D. P. Stankus, S. E. Lohse, J. E. Hutchison and J. A. Nason, Environ. Sci. Technol., 2011, 45, 3238–3244 CrossRef CAS PubMed.
- X. Lu, H. Gao, C. Li, Y.-W. Yang, Y. Wang, Y. Fan, G. Wu and J. Ma, Int. J. Pharm., 2012, 423, 195–201 CrossRef CAS PubMed.
- E. Córdova, J. Gao and G. M. Whitesides, Anal. Chem., 1997, 69, 1370–1379 CrossRef.
- B. Tah, P. Pal and G. B. Talapatra, J. Lumin., 2014, 145, 81–87 CrossRef CAS.
- R. A. Sperling and W. J. Parak, Philos. Trans. R. Soc., A, 2010, 368, 1333–1383 CrossRef CAS PubMed.
- T. H. L. Nghiem, T. H. La, X. H. Vu, V. H. Chu, T. H. Nguyen, Q. H. Le, E. Fort, Q. H. Do and H. N. Tran, Adv. Nat. Sci.: Nanosci. Nanotechnol., 2010, 1, 025009 CrossRef.
- S. M. Kelly, T. J. Jess and N. C. Price, Biochim. Biophys. Acta, Gen. Subj., 2005, 1751, 119–139 CrossRef CAS PubMed.
- P. Manavalan and W. C. Johnson, Nature., 1983, 305, 831–832 CrossRef CAS.
- A. Barzegar, A. A. Moosavi-Movahedi, S. Rezaei-Zarchi, A. A. Saboury, M. R. Ganjali, P. Norouzi, G. H. Hakimelahi and F.-Y. Tsai, Biotechnol. Appl. Biochem., 2008, 49, 203–211 CrossRef CAS PubMed.
- Q. Hua and M. A. Weiss, J. Biol. Chem., 2004, 279, 21449–21460 CrossRef CAS PubMed.
- N. O. Fischer, C. M. McIntosh, J. M. Simard and V. M. Rotello, Proc. Natl. Acad. Sci. U. S. A., 2002, 99, 5018–5023 CrossRef CAS PubMed.
- S. Chakraborty, P. Joshi, V. Shanker, Z. A. Ansari, S. P. Singh and P. Chakrabarti, Langmuir, 2011, 27, 7722–7731 CrossRef CAS PubMed.
- J. Rodríguez-Fernández, J. Pérez-Juste, F. J. G. de Abajo and L. M. Liz-Marzán, Langmuir, 2006, 22, 7007–7010 CrossRef PubMed.
- A. Neely, C. Perry, B. Varisli, A. K. Singh, T. Arbneshi, D. Senapati, J. R. Kalluri and P. C. Ray, ACS Nano, 2009, 3, 2834–2840 CrossRef CAS PubMed.
- S. D. Perrault and W. C. W. Chan, J. Am. Chem. Soc., 2009, 131, 17042–17043 CrossRef CAS PubMed.
- T. K. Sau and C. J. Murphy, J. Am. Chem. Soc., 2004, 126, 8648–8649 CrossRef CAS PubMed.
- C. Ziegler and A. Eychmüller, J. Phys. Chem. C, 2011, 115, 4502–4506 CAS.
- X. Liu, M. Atwater, J. Wang and Q. Huo, Colloids Surf., B, 2007, 58, 3–7 CrossRef CAS PubMed.
- S. Link and M. A. El-Sayed, J. Phys. Chem. B, 1999, 103, 4212–4217 CrossRef CAS.
- L. Li, Q. Mu, B. Zhang and B. Yan, Analyst, 2010, 135, 1519–1530 RSC.
- Y. Ikeda, N. Taniguchi and T. Noguchi, J. Biol. Chem., 2000, 275, 9150–9156 CrossRef CAS PubMed.
Footnote |
| † Electronic supplementary information (ESI) available. See DOI: 10.1039/c4ra17026a |
| This journal is © The Royal Society of Chemistry 2015 |