
Formal [4 + 1] cycloaddition of o-quinone methides: facile synthesis of dihydrobenzofurans†
Xiantao Leiab,
Chun-Huan Jianga,
Xiaoan Wena,
Qing-Long Xu*a and
Hongbin Sun*a
aJiangsu Key Laboratory of Drug Discovery for Metabolic Diseases, State Key Laboratory of Natural Medicines, Center of Drug Discovery, China Pharmaceutical University, 24 Tongjia Xiang, Nanjing 210009, China. E-mail: qlxu@cpu.edu.cn; hbsun2000@yahoo.com
bState Key Laboratory of Drug Research, Shanghai Institute of Materia Medica, Chinese Academy of Science, 555 Zuchongzhi Road, Shanghai 201203, China
First published on 22nd January 2015
Abstract
An efficient and straightforward method for the rapid synthesis of 2-substituted dihydrobenzofurans has been developed via reaction of sulfur ylides with o-quinone methides (o-QMs), which were generated under mild conditions. The products could be obtained in excellent yields with various kinds of sulfur ylides.
Introduction
ortho-Quinone methides (o-QMs) have been of great interest to the chemical and biological community due to their distinctive properties as Michael acceptors.1,2 Until now, various methodologies for the generation of o-QMs in situ have been developed, including tautomerization, oxidation, acid or base catalysis, thermolysis, photolysis and olefination of o-quinones.3,4 Moreover, the Rokita group reported that O-silylated phenols exposed to fluoride could also produce o-QMs under mild reaction conditions.5 In contrast to Rokita's method for generating o-QMs, other methods have several disadvantages, such as harsh reaction conditions (e.g. photolysis, high reaction temperature), long reaction time, or using transition metals as oxidants.o-QMs undergo rapid rearomatization through either Michael addition with nucleophiles, or [4 + 2] cycloaddition with dienophiles, or oxa-6π-electrocyclisation to give benzopyrans.3,6 Recently, the Scheidt and Ye groups reported NHC-catalyzed enantioselective formal [4 + 3] cycloaddition of α,β-unsaturated aldehydes with reactive o-QMs to give 2-benzoxopinones, providing a new reaction mode for o-QMs.7 On the other hand, the Zhou and Osyanin groups disclosed that o-QMs, which were generated from inaccessible substrates or in the presence of an oxidant, underwent formal [4 + 1] cycloaddition with sulfur ylides or pyridinium methylides to afford trans-2,3-dihydrobenzofurans.8 Herein, we report an efficient and straightforward method for the rapid synthesis of 2-substituted dihydrobenzofurans, which widely exist in natural products,9 via reaction of sulfur ylides with o-QMs.
Results and discussion
Our initial investigation was focused on the model reaction of O-silylated phenol 1a with sulfur ylide 2a. The reaction proceeded smoothly to provide the desired product 3a in 81% yield at 0 °C when TBAF was employed as the fluoride source (entry 1, Table 1). When the equivalent of sulfur ylide 2a was raised to 1.5 equiv., the product yield increased to 89% (entry 3, Table 1). Considering the crucial role of the fluoride source in triggering the generation of o-QMs intermediates, other fluorides such as CsF were examined. We found that CsF displayed the same reactivity with that of TBAF (entry 4, Table 1). Subsequently, the equivalent of fluoride and different temperature were examined, and it was found that the best result could be obtained at room temperature with 2.5 equiv. of TBAF (entry 8, Table 1). Finally, the influence of solvents was examined. In all solvents tested, including CH2Cl2, CHCl3, THF, Et2O, toluene, DMF, MeCN, and MeOH, the desired product was obtained in satisfactory yields (entries 8–15, Table 1), indicating a good solvent tolerance of this reaction. In terms of the product yields and operation convenience, CH2Cl2 was chosen as the best solvent.
|
||||||
|---|---|---|---|---|---|---|
| Entry | F− source | X | Y | Solvent | Temp. (°C) | Yieldb (%) |
| a 1a (0.5 mmol), 2a, solvent (5 mL), room temperature, 12 h. [TBAF (1 M in THF solution)].b Isolated yields. | ||||||
| 1 | TBAF | 1.0 | 2.5 | DCM | 0 | 81 |
| 2 | TBAF | 1.2 | 2.5 | DCM | 0 | 85 |
| 3 | TBAF | 1.5 | 2.5 | DCM | 0 | 89 |
| 4 | CsF | 1.5 | 2.5 | DCM | 0 | 85 |
| 5 | TBAF | 1.5 | 1.5 | DCM | 0 | 88 |
| 6 | TBAF | 1.5 | 1.0 | DCM | 0 | 82 |
| 7 | TBAF | 1.5 | 2.5 | DCM | −30 | 36 |
| 8 | TBAF | 1.5 | 2.5 | DCM | rt | 89 |
| 9 | TBAF | 1.5 | 2.5 | CHCl3 | rt | 86 |
| 10 | TBAF | 1.5 | 2.5 | THF | rt | 88 |
| 11 | TBAF | 1.5 | 2.5 | Et2O | rt | 86 |
| 12 | TBAF | 1.5 | 2.5 | Toluene | rt | 74 |
| 13 | TBAF | 1.5 | 2.5 | DMF | rt | 70 |
| 14 | TBAF | 1.5 | 2.5 | MeCN | rt | 77 |
| 15 | TBAF | 1.5 | 2.5 | MeOH | rt | 71 |

With the optimized reaction conditions in hand, we explored the reaction scope with using a variety of O-silylated phenols 1 and sulfur ylides 2 (Table 2). Sulfur ylide derivatives bearing electron-donating and withdrawing groups on the phenyl group were well-tolerated (3a–j), affording the corresponding products in 83–92% yields. In general, other aromatic substitutes (2-nathyl, 2-furyl, 2-thiazolyl, 2-pyridyl) derived sulfur ylides had little influence on the yields (77–89% yields, entries 12–15, Table 2). Interestingly, ester and amide derived sulfur ylides could be obtained in situ, affording the desired products in 42% and 85% yield, respectively (entries 16 and 17, Table 2). Furthermore, the 4-Br substituted O-silylated phenols 1b reacted well with 2a to afford 2-substituted dihydrobenzofuran (3q) in 90% yield (entry 18, Table 2).
|
||||
|---|---|---|---|---|
| Entry | R1 | X | R2 | Yieldb (%) |
| a 1 (0.5 mmol), 2 (0.75 mmol), TBAF (2.5 equiv., 1 M in THF solution), CH2Cl2 (5 mL), room temperature, 12 h.b Isolated yields.c Sulfonium salts was used instead of sulfur ylides, using Cs2CO3 (1.5 equiv.) as a base. | ||||
| 1 | H | Br | Ph | 89 (3a) |
| 2 | H | Cl | Ph | 92 (3a) |
| 3 | H | Br | 4-MeC6H4 | 81 (3b) |
| 4 | H | Br | 4-MeOC6H4 | 80 (3c) |
| 5 | H | Br | 3-MeOC6H4 | 91 (3d) |
| 6 | H | Br | 4-FC6H4 | 86 (3e) |
| 7 | H | Br | 2-FC6H4 | 85 (3f) |
| 8 | H | Br | 4-ClC6H4 | 92 (3g) |
| 9 | H | Br | 3-ClC6H4 | 90 (3h) |
| 10 | H | Br | 4-BrC6H4 | 83 (3i) |
| 11 | H | Br | 3-BrC6H4 | 84 (3j) |
| 12 | H | Br | 2-Naphthyl | 84 (3k) |
| 13 | H | Br | 2-Furyl | 81 (3l) |
| 14 | H | Br | 2-Thiazolyl | 95 (3m) |
| 15 | H | Br | 2-Pyridyl | 77 (3n) |
| 16c | H | Br | OEt | 42 (3o) |
| 17c | H | Br | NEt2 | 85 (3p) |
| 18 | 4-Br | Br | Ph | 90 (3q) |

We further turned our attention to the reaction with chiral sulfonium salts derived from camphor. As shown in Scheme 1, moderate enantioselectivity (63% ee) was observed in reaction of 1a with chiral sulfonium salt 4a (R′ = H),10 affording optically active amide 3p′ in only 25% yield. When using the chiral sulfonium salt 4b (R′ = Me),11 the yield of the desired product could be increased to 69%, but with the similar enantioselectivity (60% ee).
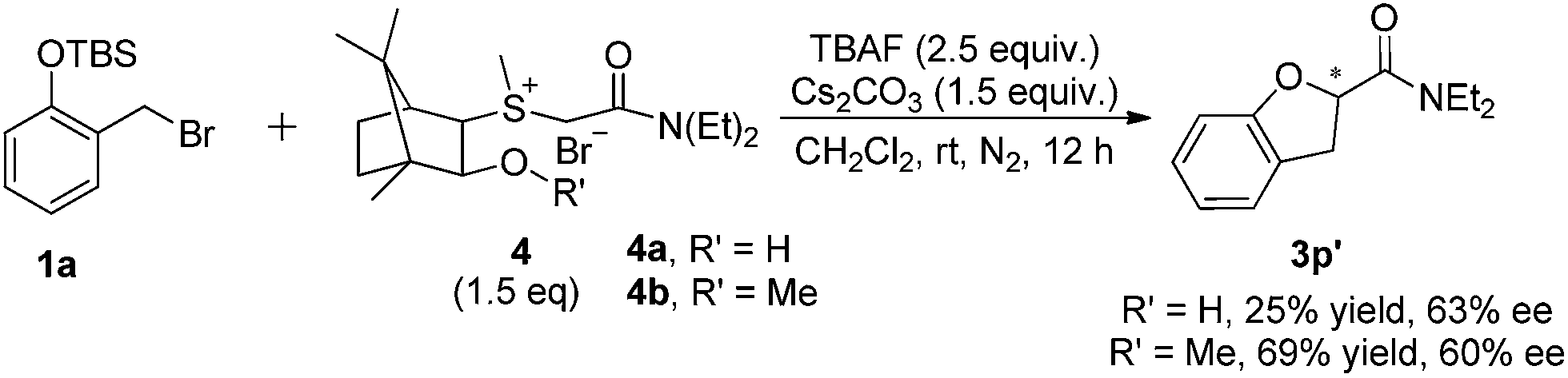 | ||
| Scheme 1 Reaction of 1a with chiral sulfonium salt 4. | ||
2-Substituted dihydrobenzofurans could be further converted into the corresponding aromatic benzofurans with using DDQ as the oxidizing agent. Thus, 2-benzoyl dihydrobenzofuran 3a was transformed smoothly to benzofuran 5a in 81% yield according to the known literature method.12 When using 2-furoyl dihydrobenzofuran 3l and 2-picolinyl dihydrobenzofuran 3n under the same conditions, the desired products 5l and 5n were obtained in moderate yields (53–56% yields), Notably, 2-thiazole formyl dihydrobenzofuran 3m was employed in the reaction, affording the product 5m with 81% yield. When the 2-substituted dihydrobenzofuran 3o and 3p was employed, the corresponding benzofuran 5o and 5p could be obtained in 75% and 69% yield, respectively (Scheme 2).
 | ||
| Scheme 2 Synthesis of benzofuran 5. | ||
A plausible mechanism for this formal [4 + 1] cycloaddition reaction is depicted in Scheme 3. We proposed that the highly reactive o-QMs A generated through desilylation/elimination reaction. Then sulfur ylides as nucleophiles attacked at the external carbon of o-QMs, affording the phenoxide B. Finally, the intermediate B lost one molecular dimethyl sulfide through nucleophilic attack, providing the 2-substituted dihydrobenzofuran.
 | ||
| Scheme 3 Proposed mechanism. | ||
Experimental section
General procedure for the reaction of O-silylated phenols and sulfur ylides
To a solution of sulfur ylides 2 (0.75 mmol, 1.5 equiv.) in dry CH2Cl2 (4 mL), a solution of O-silylated phenol 1 (0.5 mmol, 1.0 equiv.) in dry CH2Cl2 (1 mL) was added at room temperature under N2. Then TBAF (1.0 M in THF, 1.25 mL, 2.5 equiv.) was added dropwise. After the addition, the mixture was stirred at room temperature for 4 h. The reaction was concentrated under reduced pressure and purified by flash chromatography on silica gel (petroleum ether![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) :ethyl acetate = 20:1).
:ethyl acetate = 20:1).
General procedure for synthesis of 3o, 3p
A mixture of O-silylated phenol 1 (0.5 mmol, 1.0 equiv.), sulfonium salt 2 (0.75 mmol, 1.5 equiv.) and Cs2CO3 (0.75 mmol, 1.5 equiv.) in dry CH2Cl2 (5 mL) was stirred at room temperature under N2. Then TBAF (1.0 M in THF, 1.25 mL, 2.5 equiv.) was added dropwise. After the addition, the mixture was stirred at room temperature for 24 h. The reaction was concentrated under reduced pressure and purified by flash chromatography on silica gel (petroleum ether:ethyl acetate = 10:1–5:1).
Conclusions
In summary, we have developed a straightforward and efficient approach to 2-substitiuted dihydrobenzofurans through reaction of sulfur ylides with o-quinone methides that could be generated under mild conditions. In addition, chiral sulfonium salts could also be employed in this reaction, affording the desired product with moderate enantioselectivity. The products 2-substitiuted dihydrobenzofurans could be conveniently converted to 2-substituted benzofurans with using DDQ as the oxidant. Applications of this [4 + 1] cycloaddition reaction to other substrates and to the construction of biologically relevant compounds are ongoing in our laboratory.Acknowledgements
Financial support from National Natural Science Foundation of China (grants 81373303 and 81473080) and State Key Laboratory of Durg Research (SIMM1403KF-13) is gratefully acknowledged.Notes and references
- For reiews, see: (a) H. Amouri and J. Le Bras, Acc. Chem. Res., 2002, 35, 501 CrossRef CAS PubMed; (b) S. B. Ferreira, F. d. C. da Silva, A. C. Pinto, D. T. G. Gonzaga and V. F. Ferreira, J. Heterocycle. Chem., 2009, 46, 1080 CrossRef CAS; (c) Quinone Methides ed. S. E. Rokita, Wiley, New York, 2009 Search PubMed.
- (a) F. Doria, S. N. Richter, M. Nadai, S. Colloredo-Mels, M. Mella, M. Palumbo and M. Freccero, J. Med. Chem., 2007, 50, 6570 CrossRef CAS PubMed; (b) M. Di Antonio, F. Doria, S. N. Richter, C. Bertipaglia, M. Mella, C. Sissi, M. Palumbo and M. Freccero, J. Am. Chem. Soc., 2009, 131, 13132 CrossRef CAS PubMed; (c) H. Wang and S. E. Rokita, Angew. Chem., Int. Ed., 2010, 49, 5957 CrossRef CAS PubMed.
- For reviews see: (a) R. W. Van de Water and T. R. R. Pettus, Tetrahedron, 2002, 58, 5367 CrossRef CAS; (b) N. J. Willis and C. D. Bray, Chem.–Eur. J., 2012, 18, 9160 CrossRef CAS PubMed.
- For selected examples see: (a) J. Parrick, Can. J. Chem., 1964, 42, 190 CrossRef CAS; (b) D. A. Bolon, J. Org. Chem., 1970, 35, 3666 CrossRef CAS; (c) L. Jurd, Tetrahedron, 1977, 33, 163 CrossRef CAS; (d) S. R. Angle and W. Yang, J. Am. Chem. Soc., 1990, 112, 4524 CrossRef CAS; (e) R. Rodriguez, J. E. Moses, R. M. Adlington and J. E. Baldwin, Org. Biomol. Chem., 2005, 3, 3488 RSC; (f) C. D. Bray, Org. Biomol. Chem., 2008, 6, 2815 RSC; (g) C. D. Bray, Synlett, 2008, 2500 CAS; (h) S. Arumugam and V. V. Popik, J. Am. Chem. Soc., 2009, 131, 11892 CrossRef CAS PubMed; (i) N. Basarić, N. Cindro, Y. Hou, I. Žabčić, K. Mlinarić-Majerski and P. Wan, Can. J. Chem., 2011, 89, 221 CrossRef PubMed; (j) J. C. Green, S. Jiménez-Alonso, E. R. Brown and T. R. R. Pettus, Org. Lett., 2011, 13, 5500 CrossRef CAS PubMed; (k) T. P. Pathak and M. S. Sigman, J. Org. Chem., 2011, 76, 9210 CrossRef CAS PubMed; (l) C. Percivalle, A. La Rosa, D. Verga, F. Doria, M. Mella, M. Palumbo, M. Di Antonio and M. Freccero, J. Org. Chem., 2011, 76, 3096 CrossRef CAS PubMed; (m) S. Radomkit, P. Sarnpitak, J. Tummatorn, P. Batsomboon, S. Ruchirawat and P. Ploypradith, Tetrahedron, 2011, 67, 3904 CrossRef CAS PubMed; (n) N. Majumdar, K. A. Korthals and W. D. Wulff, J. Am. Chem. Soc., 2012, 134, 1357 CrossRef CAS PubMed.
- (a) S. E. Rokita, J. H. Yang, P. Pande and W. A. Greenberg, J. Org. Chem., 1997, 62, 3010 CrossRef CAS; (b) P. Pande, J. Shearer, J. H. Yang, W. A. Greenberg and S. E. Rokita, J. Am. Chem. Soc., 1999, 121, 6773 CrossRef CAS; (c) Y. Génisson, P. C. Tyler, R. G. Ball and R. N. Young, J. Am. Chem. Soc., 2001, 123, 11381 CrossRef PubMed; (d) A. F. Barrero, J. F. Q. del Moral, M. M. Herrador, P. Arteaga, M. Cortés, J. Benites and A. Rosellón, Tetrahedron, 2006, 62, 6012 CrossRef CAS PubMed.
- E. E. Weinert, R. Dondi, S. Colloredo-Melz, K. N. Frankenfield, C. H. Mitchell, M. Freccero and S. E. Rokita, J. Am. Chem. Soc., 2006, 128, 11940 CrossRef CAS PubMed.
- (a) H. Lv, W.-Q. Jia, L.-H. Sun and S. Ye, Angew. Chem., Int. Ed., 2013, 52, 8607 CrossRef CAS PubMed; (b) J. Izquierdo, A. Orue and K. A. Scheidt, J. Am. Chem. Soc., 2013, 135, 10634 CrossRef CAS PubMed.
- (a) M.-W. Chen, L.-L. Cao, Z.-S. Ye, G.-F. Jiang and Y.-G. Zhou, Chem. Commun., 2013, 49, 1660 RSC; (b) B. Wu, M.-W. Chen, Z.-S. Ye, C.-B. Yu and Y.-G. Zhou, Adv. Synth. Catal., 2014, 356, 383 CrossRef CAS; (c) V. A. Osyanin, D. V. Osipov and Y. N. Klimochkin, J. Org. Chem., 2013, 78, 5505 CrossRef CAS PubMed; (d) X.-G. Song, S.-F. Zhu, X.-L. Xie and Q.-L. Zhou, Angew. Chem., Int. Ed., 2013, 52, 2555 CrossRef CAS PubMed.
- (a) B. B. Jarvis, N. B. Pena, S. N. Comezoglu and M. M. Rao, Phytochemistry, 1986, 25, 533 CrossRef CAS; (b) L. Pieters, S. Dyck, M. Gao, R. Bai, E. Hamel, A. Vlietinck and G. Lemieère, J. Med. Chem., 1999, 42, 5475 CrossRef CAS PubMed; (c) S. Rattanaburi, W. Mahabusarakam, S. Phongpaichit and A. R. Carroll, Phytochem. Lett., 2012, 5, 18 CrossRef CAS PubMed; (d) T. Asai, D. Luo, Y. Obara, T. Taniguchi, K. Monde, K. Yamashita and Y. Oshima, Tetrahedron Lett., 2012, 53, 2239 CrossRef CAS PubMed.
- (a) Y.-G. Zhou, X.-L. Hou, L.-X. Dai, L.-J. Xia and M.-H. Tang, J. Chem. Soc., Perkin Trans. 1, 1999, 77 RSC; (b) V. K. Aggarwal, J. P. H. Charmant, D. Fuentes, J. N. Harvey, G. Hynd, D. Ohara, W. Picoul, R. Robiette, C. Smith, J. Vasse and C. L. Winn, J. Am. Chem. Soc., 2006, 128, 2105 CrossRef CAS PubMed.
- V. K. Aggarwal, G. Hynd, W. Picoul and J. L. Vasse, J. Am. Chem. Soc., 2002, 124, 9964 CrossRef CAS PubMed.
- A. Banerji and S. K. Nayak, J. Chem. Soc., Chem. Commun., 1990, 150 RSC.
- M. Uyanik, H. Okamoto, T. Yasui and K. Ishihara, Science, 2010, 328, 1376 CrossRef CAS PubMed.
- T. G. C. Bird, B. R. Brown, I. A. Stuart and A. W. R. Tyrrell, J. Chem. Soc., Perkin Trans. 1, 1983, 1831 RSC.
- Y. M. El-Garby, M. El-Kady, A. I. Essawy and A. Y. Mohamed, Indian J. Chem., Sect. B: Org. Chem. Incl. Med. Chem., 1981, 20, 747 Search PubMed.
- N. J. Lewis, D. R. Feller, G. K. Poochikian and D. T. Witiak, J. Med. Chem., 1974, 17, 41 CrossRef CAS.
- A. L. Mndzhoyan and M. A. Kaldrikyan, Izv. Akad. Nauk Arm. SSR, 1960, 13, 365 CAS.
Footnote |
| † Electronic supplementary information (ESI) available: The procedure for synthesis of O-silylated phenol 1 and the copies of spectra. See DOI: 10.1039/c4ra17003b |
| This journal is © The Royal Society of Chemistry 2015 |