DOI:
10.1039/C4RA16965D
(Paper)
RSC Adv., 2015,
5, 31375-31383
High surface area and oxygen-enriched activated carbon synthesized from animal cellulose and evaluated in electric double-layer capacitors†
Received
24th December 2014
, Accepted 24th March 2015
First published on 27th March 2015
Abstract
Crab shell, an abundant food waste and high volume organic resource, has been used to synthesize oxygen-enriched activated carbon. The thermal stability, surface area, morphology and surface chemical composition were characterized by thermogravimetric analysis, nitrogen adsorption, scanning electron microscopy, energy dispersive X-ray spectroscopy, Fourier transform infrared spectroscopy and X-ray photoelectron spectroscopy. The obtained activated carbon had a high surface area of 3442 m2 g−1, large pore volume of 2.327 cm3 g−1 and rich surface oxygen species of 18.50 at%. The cyclic voltammogram, galvanostatic charge–discharge and electrochemical impedance spectroscopy tests were performed to investigate the electrochemical properties of the resultant carbon electrodes. The specific capacitance was 280.6 F g−1 at a current density of 0.2 A g−1 and still remained as high as 233.4 F g−1 even at a high current density of 10 A g−1, indicating the great potential of crab shell-activated carbons in the development of electrode materials for high-performance supercapacitors.
1. Introduction
There are huge amount of lakes and oceans all over the world, therefore the production of crabs is enormous per year.1 Crab shells (CS) are one of the most common solid wastes in seafood industries. Based on up-to-date data, it is estimated that the production of crab shell is over millions of tons annually and approximately 85% of crab shells are ultimately disposed of in landfill, resulting in a serious environmental problem. Owing to the strict environmental regulations, how to reuse this solid waste in an appropriate manner and also create possible economic value to the industries is significant. The raw crab shells are mainly composed of 40–60% calcium carbonate, 20–27% chitin, 11–29% protein, 3–5% magnesium carbonate and some lipids as dry weight basis.2–4 Chitin, the second most abundant polymer in nature, can be extracted from the shells by using very simple chemical treatments. Chitin is a modified polysaccharide with a strictly hierarchical structure, which is consisted of the units of N-acetylglucosamine.5 Or rather, chitin could be regarded as one hydroxyl group on per monomer of cellulose replaced by an acetyl amine group. Chitin, also called animal cellulose, possess abundantly available carbon content and various surface chemical groups. In addition, chitin in crab shell is often associated with various types of proteins or lipids, which could also be employed as carbon precursor.
Up to now, most researches have been only focused on the reutilization of crab shells or chitin from crab shells as bioadsorbents to remove heavy metals.6–9 Activated carbons (ACs) with high surface area, large pore volume and hierarchical microporous/mesoporous structure are widely used in various fields, including adsorption,10 gas separation/storage,11,12 catalysis supporter13 and electric double-layer capacitors (EDLCs).14 However, the commercial ACs derived from relatively expensive and non-renewable starting materials such as coals, are high cost and unjustified to the pollution control applications. Recently, various types of wastes have proven to be good candidates for the synthesis of ACs, such as nut shell,15 sludge,16 alkali lignin,17 lotus stalk,18 coal tar pitch,19 rice husk20 and Enteromorpha prolifera.21 Animal cellulose from crab shell wastes could also be an available raw material for the synthesis of ACs.
According to literature reviews, no research has been reported on the preparation of low-cost activated carbon from animal cellulose of crab shells. Therefore, the main objective of this study was to explore the potential of crab shell wastes in the synthesis of activated carbon with high surface area using potassium hydroxide as activating agent. The physical/chemical properties and electrochemical performances of the obtained ACs were also investigated.
2. Experimental
2.1. Preparation of activated carbon
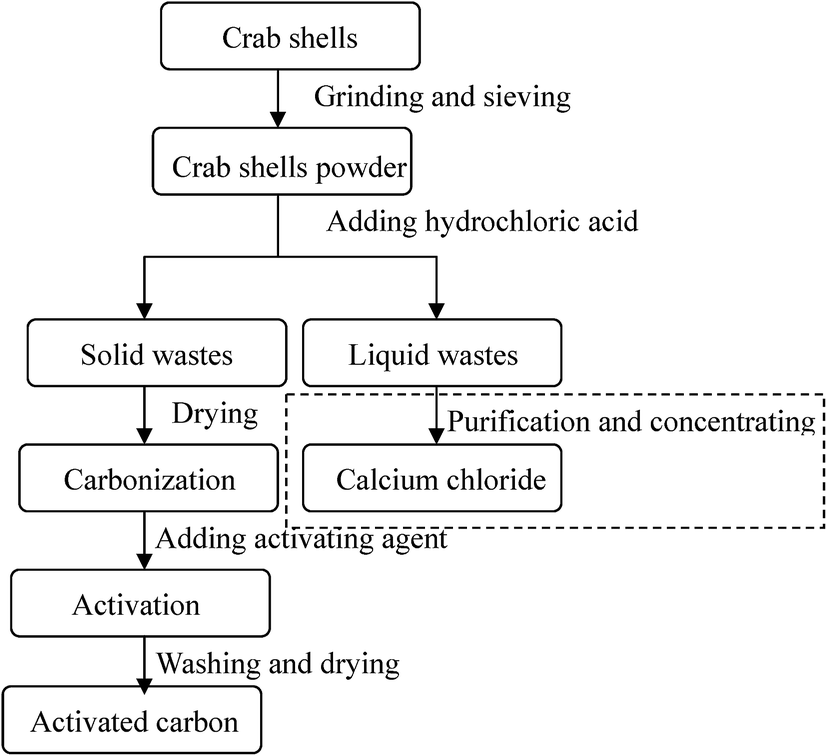
Raw crab shell wastes were collected from a crabmeat processing factory in Hebei, China. The shells were sun-dried for several days and mechanically ground to particles in size range of 0.074–0.15 mm. As shown in Fig. 1 and 2, the formation process of the ACs from crab shells was carried out on a simple and reproducible route. Firstly, the raw shell was treated with 1 N HCl at a solid–liquid ratio of 100 g L−1 for 6 h followed by rinsing with distilled water several times. This pre-treatment procedure was performed to guarantee the removal of excess minerals, like calcium carbonate or calcium phosphate, on the shell surface.22,23 The obtained acidified crab shell (ACS) was mainly composed of chitin and protein. Then, the ACS was dried in an oven at 60 °C for 24 h and the weight loss in this process was found to be approximately 55 ± 1%. Secondly, the ACS was carbonized at 500 °C for 90 min. The obtained char was denoted as CCS. The char yield in the carbonation process was 28 ± 0.5%. Then the CCS was mixed with potassium hydroxide powder. The mixture was transferred to a tube furnace and pyrolyzed under nitrogen flow at 800 °C for 60 min. After cooling under nitrogen protection, the sample was washed repeatedly with HCl solution and distilled water to remove residual alkali and inorganic impurities. Finally, the product was dried at 105 °C overnight and ground into powders. The obtained product was designated as CSAC-x (x = 1, 2, 3, 4 and 5), where x represented the weight ratio between potassium hydroxide and char. The carbon yield in the activation process was 24.5 ± 1%. This preparation process was referred from our previous work.21 The synthesis process was carried out in triplicate and the average result was obtained.
 |
| | Fig. 1 Flow diagram for the synthesis of activated carbons from crab shell wastes. | |
 |
| | Fig. 2 Mechanism diagram for the synthesis of activated carbons from crab shell wastes. | |
2.2. Characterization
Thermal gravimetric analysis (TGA) was performed using a thermogravimetric analyzer (SHI-MADZU, TGA-50). Each analysis was carried out at a 100 mL min−1 N2 rate with a heat rate of 10 °C min−1 from room temperature to 900 °C. The pore size distribution and surface area of the ACs were determined at 77 K using a surface area analyzer (JW-BK122W). The surface physical morphology and mineral components of the ACs were examined by using a scanning electron microscopy (JEOL, JSM 7600F) and an energy dispersive X-ray spectroscopy (Oxford INCA sightX). Fourier transform infrared spectroscopy (NICOLET 6700) and X-ray photoelectron spectroscopy (ESCALAB 250) tests were carried out to investigate the chemical properties of the samples.
2.3. Electrochemical measurements
All electrochemical characterizations of the CSAC-3 based electrodes were tested in a symmetrical two-electrode configuration with 6 M KOH as electrolyte using an electrochemical workstation (PARSTAT2273). To make a work electrode, the active CSAC-3 powder, acetylene black and polytetrafluoroethylene (PTFE) were mixed in ethanol with a weight ratio of 85![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) :15:5. The mixture was coated onto nickel foam like a sandwich with an area of 1 cm2 and pressed under a pressure of 10 MPa. The electrodes were dried in a vacuum oven at 100 °C overnight. Approximately 2 mg of active material was loaded in each electrode, and each electrode layer thickness was 50 μm. Electrochemical performances of the prepared carbon electrodes were evaluated by cyclic voltammetry (CV), galvanostatic charge–discharge (GCD) and electrochemical impedance spectroscopy (EIS). The potential window of CV curve was set to be 0–1.0 V. In GCD test, different current densities from 0.2 to 10 A g−1 were employed to evaluate the charge–discharge performance of the samples. The EIS test was performed in the frequency ranges of 100 kHz to 100 mHz at an open circuit potential of 10 mV. The specific capacitance based on CV and GCD measured was calculated using the following equations:
:15:5. The mixture was coated onto nickel foam like a sandwich with an area of 1 cm2 and pressed under a pressure of 10 MPa. The electrodes were dried in a vacuum oven at 100 °C overnight. Approximately 2 mg of active material was loaded in each electrode, and each electrode layer thickness was 50 μm. Electrochemical performances of the prepared carbon electrodes were evaluated by cyclic voltammetry (CV), galvanostatic charge–discharge (GCD) and electrochemical impedance spectroscopy (EIS). The potential window of CV curve was set to be 0–1.0 V. In GCD test, different current densities from 0.2 to 10 A g−1 were employed to evaluate the charge–discharge performance of the samples. The EIS test was performed in the frequency ranges of 100 kHz to 100 mHz at an open circuit potential of 10 mV. The specific capacitance based on CV and GCD measured was calculated using the following equations:| |
 | (1) |
| |
 | (2) |
where I is the current (A), u is the potential (V), v is the voltage scan rate (mV s−1), m is the weight of the active material in each electrode, ΔV is the total potential difference (V) and Δt is the discharge time (s).
3. Results and discussion
3.1. Thermal analysis
TGA analysis can provide useful information on thermal behavior of materials through the study of characteristic decomposition patterns or degradation mechanisms. Hence, TGA coupled with DTG analysis were employed in this study to gain suitable temperature range for the carbonization and activation process. Fig. 3 exhibits the TGA–DTG curves for both acidified crab shell and carbonized crab shell impregnated with KOH. Both samples display three steps for weight loss, yet remarkable variations could be discovered in the band locations and intensities. Fig. 3a shows that the decomposition of the acidified crab shell was consisted of three stages, namely dehydration, devolatilization and decomposition of carbonaceous substance, which was different from pure chitin or protein.24,25 This result demonstrated that the acidified crab shell was a combination of chitin, protein, lipid, ash and moisture, which was in agreement with the previous study.23 Furthermore, the shape of TGA curve of acidified crab shell resembled like that of cellulose, implying that the obtained acidified material could be used as a promising carbon precursor.26,27 The first mass loss of 11.44% at 30–200 °C was caused by the evaporation of water molecules, while the second mass loss observed in the temperature range of 200–408 °C was a result of the decomposition of chitin, protein or lipid molecules. Approximately 53.03% weight of the sample was lost during this stage with two significant DTG peaks at about 317.5 and 384.6 °C, which were similar to previous studies.24,25,28 In the third stage, from 408 to 800 °C, the sample release 15.77% volatile substances and a char residue of 19.76% was gained. According to the above analysis, the carbonization temperature was selected at 500 °C, because large proportion of dehydration and devolatilization of raw materials occurred in this stage.
 |
| | Fig. 3 TGA and DTG curves for the pyrolysis of (a) acidized crab shell and (b) carbonized char impregnated with potassium hydroxide. | |
On the other hand, the raw precursor could be eroded by potassium hydroxide and produced pores in the following aspects. (a) The primary surface dehydration occurred, which could be deduced from the DTG peak at 77.2 °C (Fig. 3b). (b) The secondary evaporation of internal water and partial polymerization of the raw precursor were deduced from the DTG peak at 209.8 °C. (c) The raw precursor transformed to char through aromatization reaction, which presented a high weight loss between the temperature 100 and 750 °C. During this stage, the metallic potassium coupled with some carbonates were produced to yield abundant fine pores through intercalating to the carbon matrix.29 (d) The progressive weight loss above 750 °C was attributed to the decomposition of potassium carbonate into CO2. Therefore, the activation temperature was set at 800 °C to ensure good development of pores.
3.2. Pore and microstructure characterization
It can be seen from Table S1† that raising the impregnation ratio from 1 to 3 lead to an increase in BET surface area from 1782 to 3442 m2 g−1, while further raising the impregnation ratio from 3 to 5 resulted in a decrease in BET surface area from 3442 to 2188 m2 g−1. CSAC with the highest surface area was obtained at the impregnation ratio of 3. Therefore, CSAC-3 was regarded as a typical sample for the following investigation. As shown in Fig. 4a, the N2 adsorption–desorption isotherms of CSAC-3 presented a type I shape with somewhat type IV character. The isotherms showed a significant adsorption of nitrogen at the relative pressure below 0.1, indicating the presence of micropores. A slight hysteresis loop (or capillary condensation) occurred at the medium relative pressure of 0.4–0.7, implying that CSAC-3 possessed some mesopores. And the hysteresis loop belonged to H3/H4, which was associated to the existence of slit-liked pores.30,31 The size of micropores for CSAC-3 displayed a multimodal distribution nature with four different maxima at 0.55, 0.62, 0.74 and 0.83 nm (Fig. 4b). All textural parameters of CS, ACS, CCS and CSAC-3 are presented in Table 1. The acidification and carbonization processes led to slight change to the surface area and pore volume for raw precursor. However, the surface area and pore volume increased to 3442 m2 g−1 and 2.327 cm3 g−1 after activation. Compared with some previous studies,32–35 ACs prepared from crab shell exhibited higher surface area than that of other raw precursors, such as sugarcane molasses, sunflower seed shell, rice straw and Argania spinosa seed shells. The average pore diameter for CSAC-3 was 2.704 nm and the ratio of mesopores/total pores reached 90.46%. As displayed in Fig. 4b, most pores were intensively distributed in the pore range of 2–4 nm. According to the previous study, large surface area and high proportion of mesopores for ACs facilitated the access of electrolyte ions into the pores, contributing to good electrochemical performances in EDLCs.36,37
 |
| | Fig. 4 (a) N2 sorption–desorption isothermals and (b) pore size distribution curves (inset: micropore size distribution) of the prepared carbons. | |
Table 1 Porous structure parameters of crab shell, acidized crab shell, carbonized crab shell and crab shell resulting activated carbon
| Parameters |
CS |
ACS |
CCS |
CSAC-3 |
| BET surface area (m2 g−1) |
1.817 |
5.061 |
10.45 |
3442 |
| Total pore volume (cm3 g−1) |
0.030 |
0.017 |
0.033 |
2.327 |
| t-method micropore volume (cm3 g−1) |
0.000 |
0.000 |
0.000 |
0.642 |
| BJH method mesopore volume (cm3 g−1) |
0.033 |
0.019 |
0.036 |
2.105 |
| Average pore diameter (nm) |
65.479 |
13.753 |
12.744 |
2.704 |
Scanning electron microscopy equipped with EDX was carried out to analyze the morphologies and components on the surface of CS, ACS, CCS and CSAC-3. In EDX analysis (Fig. 5), strong Ca peaks could be observed on virgin crab shell surface, which confirmed that the shells contained calcium carbonate. Other peaks corresponding to Na, Mg, P, Cl, K, Cu, Mn and Pb were also recorded in the EDX spectrum (Table S2†). It is interesting to find that the calcium peaks decreased remarkably after acidification, implying that the calcium leached out from the shells successfully through demineralization. SEM images showed that the surface of virgin crab shell display lamellar-like structure with many straightly tunnel-like and striped trench (Fig. 6a and S1a†). After acidification treatment, the ACS surface exhibited apparent microfibrillar crystalline structure in a sequence of hierarchical layer patterns, indicating that the reserved main constituents contained chitin (Fig. 6b and S1b†). Other previous researchers also found that the purified crab shell displayed similar microfibrillar structure.5,38 After carbonization, the surface became smooth with plate-like stripes (Fig. 6c and S1c†). After activation, the surface of the resultant sample presented abundantly flower-like or honeycomb-like 3-D pore network skeleton (Fig. 6d and S1d†).
 |
| | Fig. 5 EDX spectrum (a) raw crab shell and (b) acidized crab shell. | |
 |
| | Fig. 6 Scanning electron microscopy image of (a) raw crab shell, (b) acidized crab shell, (c) carbonized crab shell and (d) crab shell activated carbon (×20000). | |
3.3. Chemical characterization
The electrochemical behaviors of EDLCs not only rely on the physical properties but also on surface chemical characteristics, hence FTIR and XPS were used in this study. Fig. 7 revealed five different characteristic bands for CS, ACS, CCS and CSAC-3. These bands were: 3425 cm−1 (O–H stretching vibration), 2910 cm−1 (C–H stretching), 1648 cm−1 (C![[double bond, length as m-dash]](https://www.rsc.org/images/entities/char_e001.gif) O secondary amide stretching), 1425 cm−1 (C–H deformation) and 1060 cm−1 (O–C–O asymmetric stretching in phase ring),24,39 which became weaken, shifted or disappeared for CSAC-3, indicating that the hydrogen element was removed to a large extent due to the dehydration or pyrolyzation reaction during activation process. The surface elemental compositions of the prepared carbon were further evaluated by XPS, listed in Fig. S2.† It can be seen that the C1s spectrum could be deconvoluted into three components, corresponding to: graphite type (284.6 eV), carbon in phenolic, alcoholic, etheric groups (285.6 eV) and carbon in carboxyl or ester groups (288.7 eV).39,40 The O1s spectra could be resolved into three individual component peaks: oxygen in hydroxyl or metal oxides (530.6 eV), oxygen doubly bonded to carbon (532.4 eV) and oxygen singly bonded to carbon in aromatic rings, phenols and ethers (533.1 eV).40,41 According to the area-simulating curve, the relative percentage of each component was calculated and summarized in Table 2. The relative content of oxygen in aromatic rings, phenols and ethers was 7.60%, which was very high. According to the previous studies, CO-type functional groups on the surface of ACs, including hydroxyl, quinine, phenols, ethers and carbonyl groups, had a positive contribution to the specific capacity of carbon electrodes.42–44 Since large numbers of O-enriched functional groups could not only promote the wettability of carbon material, thus favouring the electrolyte ions to contact with the pores, but also provide an additional pseudocapacitance due to a Faradic process involving the oxygen groups.42,43
O secondary amide stretching), 1425 cm−1 (C–H deformation) and 1060 cm−1 (O–C–O asymmetric stretching in phase ring),24,39 which became weaken, shifted or disappeared for CSAC-3, indicating that the hydrogen element was removed to a large extent due to the dehydration or pyrolyzation reaction during activation process. The surface elemental compositions of the prepared carbon were further evaluated by XPS, listed in Fig. S2.† It can be seen that the C1s spectrum could be deconvoluted into three components, corresponding to: graphite type (284.6 eV), carbon in phenolic, alcoholic, etheric groups (285.6 eV) and carbon in carboxyl or ester groups (288.7 eV).39,40 The O1s spectra could be resolved into three individual component peaks: oxygen in hydroxyl or metal oxides (530.6 eV), oxygen doubly bonded to carbon (532.4 eV) and oxygen singly bonded to carbon in aromatic rings, phenols and ethers (533.1 eV).40,41 According to the area-simulating curve, the relative percentage of each component was calculated and summarized in Table 2. The relative content of oxygen in aromatic rings, phenols and ethers was 7.60%, which was very high. According to the previous studies, CO-type functional groups on the surface of ACs, including hydroxyl, quinine, phenols, ethers and carbonyl groups, had a positive contribution to the specific capacity of carbon electrodes.42–44 Since large numbers of O-enriched functional groups could not only promote the wettability of carbon material, thus favouring the electrolyte ions to contact with the pores, but also provide an additional pseudocapacitance due to a Faradic process involving the oxygen groups.42,43
 |
| | Fig. 7 FTIR spectra of (a) raw crab shell, (b) acidized crab shell, (c) carbonized crab shell and (d) crab shell activated carbon. | |
Table 2 Relative content of the surface functional groups determined from the XPS analysis
| Peaks no. |
Binding energy (eV) |
Surface group |
Assignment |
Relative content (%) |
| C1s |
Peak 1 |
284.6 |
C |
Graphitic carbon |
35.92 |
| Peak 2 |
285.6 |
C–O– |
Phenolic, alcoholic, etheric |
20.10 |
| Peak 3 |
288.7 |
COO |
Carboxyl or ester |
24.21 |
| O1s |
Peak 1 |
530.6 |
O/OH– |
Hydroxyl or metal oxides |
0.63 |
| Peak 2 |
532.4 |
CO |
Oxygen doubly bonded to carbon |
10.27 |
| Peak 3 |
533.1 |
C–O– |
Aromatic rings, in phenols and ethers |
7.60 |
| N1s |
Peak 1 |
400.2 |
C–N–C |
Pyrrolic nitrogen, pyridones or a mixture of both |
1.27 |
3.4. Electrochemical behavior
Fig. 8a presents the CV curves of CSAC-3 electrodes at various scan rates of 10–100 mV s−1 in 6 M KOH aqueous electrolyte. The CSAC-3 electrodes displayed good rectangular shapes, exhibiting almost mirror images with reference to the zero-current line, which was a typical characteristic of the EDLCs. Furthermore, the rectangular degree of the curves presented the ion diffusion rate into the pores of the electrodes, and the higher rectangular degree reflects bigger ion diffusion rate.36 Hence, the high rectangular degree of CV curves at high scan rates in this study corresponded to the good ion transfer properties of the CSAC-3 electrode materials. It is well-known that the RC time constant (τ) of the electrode is an important influence factor on the voltammogram shape. The larger the constant time is, the longer the transient part (less steepness of the CV curves at the switching potential) will be, which signifies worse collapse of the rectangular profiles.37,45,46 In addition, this phenomenon usually becomes more prominent at higher scan rates. In this study, the CV curves still maintained a good box-like shape at high scan rates, which implied that the carbon electrode had small constant time as a desired capacitor with an excellent capacitive ability. The RC time constant is only 5.01 s, which indicated that the CSAC-3 electrode was suitable for higher power delivery.
 |
| | Fig. 8 (a) Cyclic voltammograms at different scan rates, (b) charge–discharge curves and (c) the specific capacitance at various current densities of CSAC-3 electrodes. | |
The galvanostatic charge–discharge measurement at different current densities was further employed to explore the electrochemical behaviors of the electrode materials. All galvanostatic charge–discharge curves show almost isosceles triangular in shape even at high current density of 2.5 A g−1 (Fig. 8b), suggesting an ideal electrochemical reversibility of the electrodes. Additionally, the specific capacitance of CSAC-3 electrodes was 280.6 F g−1 at a current density of 0.2 A g−1 and still remained as high as 233.4 F g−1 when the current density increased up to 10 A g−1 (Fig. 8c), which is comparable to those reported elsewhere.19,20,37,45 The decrease of the capacitance mainly derived from ohmic drop or high polarization of electrode at high current density, which could result from the weakening accessibility of ions into the pores of electrode matrices with the increasing current density.36 The high capacitance retention of 83% reflected an excellent rate capability of the electrodes.
To evaluate the effects of frequency on the power performance of CSAC-3 electrodes, the prepared carbon electrodes were investigated by electrochemical impedance spectroscopy. As shown in Fig. 9a, the Nyquist plot consist of three parts: (a) a semicircle at high frequency region, which was correlated to the resistance of the CSAC-3 electrode itself as well as the contact resistance between CSAC-3 electrode and current collector. During this stage, the value of capacitance was almost zero and the system behaved like a pure resistor (Fig. 9b). Because scarcely any charge transfer complexes could overcome the activation energy to move with the quick changes of the potential; (b) a 45° Warburg line at middle frequency region served as a knee or transition between the semicircle and vertical line; (c) a vertical line at low frequency region implied a pure capacitive electric double layer capacitor behavior.31,36 The data of Nyquist curve was fitted by using the ZSimpWin software based on the equivalent circuit (Fig. 9a inset). The equivalent series resistance of the electrode was 0.194 Ω, which made it possible for high power performance. The excellent electrochemical performance of CSAC-3 electrode may attribute to the characteristics of the crab shell carbon materials. Firstly, large specific surface area provided sufficient electrode–electrolyte interface sites for charge storage. Secondly, dominant mesopores in the porous carbon material resulted in low inner-pore ion-transport resistance and diffusion distance, which facilitated the electrolyte ion to transfer and access into the internal pores quickly.37,47 Thirdly, the presence of abundant oxygen-contained groups enhanced the surface wettability of the electrode surface, affording more exposed surface for charge accommodation.
 |
| | Fig. 9 (a) Nyquist impedance plots and normalized real and imaginary part capacitance of the CSAC-3 electrodes. | |
4. Conclusions
Novel oxygen-rich activated carbons were fabricated by KOH activation using animal cellulose withdrawn from crab shell wastes as precursor and applied as electrode materials. The obtained carbon materials presented outstanding performance as an EDLC electrode owing to large specific surface area of 3442 m2 g−1, high mesopores ratio of 90.46% and high oxygen content of 18.50%. The advantages of the raw material were: (1) abundantly available and cheap; (b) instinctively high oxygen content; (c) simple and cost-effective synthesis; (d) good electrochemical performance. This investigation provides an interesting and promising candidate for supercapacitor industry.
References
- D. S. Kim, Bioresour. Technol., 2004, 94, 345–348 CrossRef CAS PubMed.
- K. Vijayaraghavan, H. Y. N. Winnie and R. Balasubramanian, Desalination, 2011, 266, 195–200 CrossRef CAS PubMed.
- S. Lu, S. W. Gibb and E. Cochrane, J. Hazard. Mater., 2007, 149, 208–217 CrossRef CAS PubMed.
- K. Vijayaraghavan, K. Palanivelu and M. Velan, Bioresour. Technol., 2006, 97, 1411–1419 CrossRef CAS PubMed.
- M.-T. Yen, J.-H. Yang and J.-L. Mau, Carbohydr. Polym., 2009, 75, 15–21 CrossRef CAS PubMed.
- E. I. Cadogan, C.-H. Lee, S. R. Popuri and H.-Y. Lin, Int. Biodeterior. Biodegrad., 2014, 95, 232–240 CrossRef CAS PubMed.
- K. Vijayaraghavan and R. Balasubramanian, Chem. Eng. J., 2010, 163, 337–343 CrossRef CAS PubMed.
- I. B. Rae, S. W. Gibb and S. Lu, J. Hazard. Mater., 2009, 164, 1601–1604 CrossRef CAS PubMed.
- P. X. Pinto, S. R. Al-Abed and D. J. Reisman, Chem. Eng. J., 2011, 166, 1002–1009 CrossRef CAS PubMed.
- M. Houari, B. Hamdi, O. Bouras, J.-C. Bollinger and M. Baudu, Chem. Eng. J., 2014, 255, 506–512 CrossRef CAS PubMed.
- K. Kante, C. Nieto-Delgado, J. R. Rangel-Mendez and T. J. Bandosz, J. Hazard. Mater., 2012, 201–202, 141–147 CrossRef CAS PubMed.
- N. Ferrera-Lorenzo, E. Fuente, I. Suárez-Ruiz and B. Ruiz, Chem. Eng. J., 2014, 250, 128–136 CrossRef CAS PubMed.
- L. L. Bo, Y. B. Zhang, X. Quan and B. Zhao, J. Hazard. Mater., 2008, 153, 1201–1206 CrossRef CAS PubMed.
- S. Kumagai, M. Sato and D. Tashima, Electrochim. Acta, 2013, 114, 617–626 CrossRef CAS PubMed.
- A. C. Martins, O. Pezoti, A. L. Cazetta, K. C. Bedin, D. A. S. Yamazaki, G. F. G. Bandoch, T. Asefa, J. V. Visentainer and V. C. Almeida, Chem. Eng. J., 2015, 260, 291–299 CrossRef CAS PubMed.
- P. Hadi, M. Xu, C. Ning, C. Sze Ki Lin and G. McKay, Chem. Eng. J., 2015, 260, 895–906 CrossRef CAS PubMed.
- S. Hu, S. Zhang, N. Pan and Y.-L. Hsieh, J. Power Sources, 2014, 270, 106–112 CrossRef CAS PubMed.
- H. Liu, W. Liu, J. Zhang, C. Zhang, L. Ren and Y. Li, J. Hazard. Mater., 2011, 185, 1528–1535 CrossRef CAS PubMed.
- X. He, H. Zhang, H. Zhang, X. Li, N. Xiao and J. Qiu, J. Mater. Chem. A, 2014, 2, 19633 CAS.
- X. He, P. Ling, J. Qiu, M. Yu, X. Zhang, C. Yu and M. Zheng, J. Power Sources, 2013, 240, 109–113 CrossRef CAS PubMed.
- Y. Gao, W. Zhang, Q. Yue, B. Gao, Y. Sun, J. Kong and P. Zhao, J. Power Sources, 2014, 270, 403–410 CrossRef CAS PubMed.
- C. H. Niu, B. Volesky and D. Cleiman, Water Res., 2007, 41, 2473–2478 CrossRef CAS PubMed.
- H. Niu and B. Volesky, Hydrometallurgy, 2006, 84, 28–36 CrossRef CAS PubMed.
- M. Kaya, O. Seyyar, T. Baran, S. Erdogan and M. Kar, Int. J. Biol. Macromol., 2014, 65, 553–558 CrossRef CAS PubMed.
- K. Kebelmann, A. Hornung, U. Karsten and G. Griffiths, Biomass Bioenergy, 2013, 49, 38–48 CrossRef CAS PubMed.
- P. R. Sharma and A. J. Varma, Carbohydr. Polym., 2014, 114, 339–343 CrossRef CAS PubMed.
- Q. Liu, Z. Zhong, S. Wang and Z. Luo, J. Anal. Appl. Pyrolysis, 2011, 90, 213–218 CrossRef CAS PubMed.
- M. Kaya, S. Erdogan, A. Mol and T. Baran, Int. J. Biol. Macromol., 2015, 72, 797–805 CrossRef CAS PubMed.
- A.-N. A. El-Hendawy, Appl. Surf. Sci., 2009, 255, 3723–3730 CrossRef CAS PubMed.
- M. E. Fernandez, G. V. Nunell, P. R. Bonelli and A. L. Cukierman, Ind. Crops Prod., 2014, 62, 437–445 CrossRef CAS PubMed.
- Q. Wang, J. Yan, Y. Wang, T. Wei, M. Zhang, X. Jing and Z. Fan, Carbon, 2014, 67, 119–127 CrossRef CAS PubMed.
- A. H. Basta, V. Fierro, H. El-Saied and A. Celzard, Bioresour. Technol., 2009, 100, 3941–3947 CrossRef CAS PubMed.
- A. Elmouwahidi, Z. Zapata-Benabithe, F. Carrasco-Marin and C. Moreno-Castilla, Bioresour. Technol., 2012, 111, 185–190 CrossRef CAS PubMed.
- X. Li, W. Xing, S. Zhuo, J. Zhou, F. Li, S. Z. Qiao and G. Q. Lu, Bioresour. Technol., 2011, 102, 1118–1123 CrossRef CAS PubMed.
- J. Sreńscek-Nazzal, W. Kamińska, B. Michalkiewicz and Z. C. Koren, Ind. Crops Prod., 2013, 47, 153–159 CrossRef PubMed.
- M. Wu, P. Ai, M. Tan, B. Jiang, Y. Li, J. Zheng, W. Wu, Z. Li, Q. Zhang and X. He, Chem. Eng. J., 2014, 245, 166–172 CrossRef CAS PubMed.
- W. Zhang, Z.-H. Huang, G. Cao, F. Kang and Y. Yang, J. Power Sources, 2012, 204, 230–235 CrossRef CAS PubMed.
- O. C. Wilson, A. Gugssa, P. Mehl and W. Anderson, Mater. Sci. Eng., C, 2012, 32, 78–82 CrossRef CAS PubMed.
- H. Liu, X. Wang, G. Zhai, J. Zhang, C. Zhang, N. Bao and C. Cheng, Chem. Eng. J., 2012, 209, 155–162 CrossRef CAS PubMed.
- A. P. Terzyk, Colloids Surf., A, 2001, 77, 23–45 CrossRef.
- S. Altenor, B. Carene, E. Emmanuel, J. Lambert, J.-J. Ehrhardt and S. Gaspard, J. Hazard. Mater., 2009, 165, 1029–1039 CrossRef CAS PubMed.
- V. Ruiz, C. Blanco, E. Raymundo-Piñero, V. Khomenko, F. Béguin and R. Santamaría, Electrochim. Acta, 2007, 52, 4969–4973 CrossRef CAS PubMed.
- M. J. Bleda-Martínez, J. A. Maciá-Agulló, D. Lozano-Castelló, E. Morallón, D. Cazorla-Amorós and A. Linares-Solano, Carbon, 2005, 43, 2677–2684 CrossRef PubMed.
- H. Oda, A. Yamashita, S. Minoura, M. Okamoto and T. Morimoto, J. Power Sources, 2006, 158, 1510–1516 CrossRef CAS PubMed.
- H.-Q. Li, Y.-G. Wang, C.-X. Wang and Y.-Y. Xia, J. Power Sources, 2008, 185, 1557–1562 CrossRef CAS PubMed.
- S. Yoon, J. Lee, T. Hyeon and S.-M. Oh, J. Electrochem. Soc., 2000, 147(7), 2507–2512 CrossRef CAS PubMed.
- J. Wang, M.-M. Chen, C.-Y. Wang, J.-Z. Wang and J.-M. Zheng, J. Power Sources, 2011, 196, 550–558 CrossRef CAS PubMed.
Footnote |
| † Electronic supplementary information (ESI) available. See DOI: 10.1039/c4ra16965d |
|
| This journal is © The Royal Society of Chemistry 2015 |
Click here to see how this site uses Cookies. View our privacy policy here.