
One-step synthesis of a copper-based metal–organic framework–graphene nanocomposite with enhanced electrocatalytic activity†
Juan Yang,
Faqiong Zhao* and
Baizhao Zeng
Key Laboratory of Analytical Chemistry for Biology and Medicine (Ministry of Education), College of Chemistry and Molecular Sciences, Wuhan University, Wuhan, Hubei 430072, China. E-mail: fqzhao@whu.edu.cn
First published on 13th February 2015
Abstract
A copper-based metal–organic framework–graphene nanocomposite (Cu-MOF–GN) (i.e. Cu3(BTC)2, BTC = 1,3,5-benzene-tricarboxylate) was prepared by a facile one-step method for the first time. Unlike the conventional strategies, in this procedure graphene oxide was reduced to GN by an endogenous reducing agent produced by dimethylformamide, which was used as solvent in the synthesis of Cu-MOF. The nanocomposite exhibited high stability due to the hydrogen bonding, π–π stacking and Cu–O coordination between Cu-MOF and GN. Owing to the synergetic effect of Cu-MOF and GN, the Cu-MOF–GN nanocomposite showed high electrocatalytic activity. When it was used for constructing H2O2 and ascorbic acid sensors, it presented good performance. Thus, the Cu-MOF–GN nanocomposite has potential applications in the electrochemical field.
1. Introduction
As a leading class of porous materials, metal–organic frameworks (MOFs), formed by metal ions or metal ion clusters and bridging organic linkers, have many unique properties such as a crystalline ordered structure, large surface area, high thermal stability, extra high porosity and open metal sites.1–5 Over the past decades, many researchers have focused on their applications in gas storage and separation.6–9 Recently, the utilizations of MOFs in electrochemical field attracted tremendous attention.10–15 This is due to the electroactivity of metal ions and ligands used in MOFs, which could provide a pathway for electrons.16–20 For example, Zhang et al. reported the electrocatalytic oxidation of H2O2 at a Cu-based MOF modified electrode, which was related to the reversible redox reaction of CuIII(OH)-MOF/CuII(H2O)-MOF couple.21 Jia et al. discussed the electrocatalytic cycle mechanism of a Cu-based MOF for the dimethyl carbonate synthesis.22 A Fe-based MOF modified platinum disk electrode for the oxidation of hydroxide to O2 was developed by Babu et al.,23 concerning the reversible reduction of Fe(III).However, the practical electrochemical applications of single-phase MOF materials are still restricted due to their inferior electronic conductivity, low mechanical stability and poor electrocatalytic ability.24 To solve this problem, an efficient strategy is to introduce some highly conductive and mechanically durable materials into MOFs, such as metal nanocrystals, conductive polymers, carbon nanostructures.25–27 Obviously, the MOF-carbon composites are preferred among them because of the high conductivity, low cost, high catalytic activity and stability of carbon materials. Some researchers have made efforts in this regard. For instance, Cu-MOF–macroporous carbon (MPC)/GCE,24 Cu-MOF–multiwalled carbon nanotube (MWCNTs)/GCE,28 Fe-MOF–pyridine functionalized graphene (G-dye)/GCE,29 carbon black–Fe–MOF/GCE30 and some carbon paste electrodes31,32 were reported as electrochemical sensor. Deservedly, due to the unique characteristics of graphene (GN),33 the MOF–GN composites also have been widely studied. Nevertheless, these materials suffer from the complicated synthesis process, mainly including two steps: the reduction of graphene oxide (GO) with proper reducing reagents and the binding of MOF and GN. For example, Wang et al.34 reported a Cu-MOF–EGR (electrochemically reduced graphene)/GCE electrode for acetaminophen (ACOP) and dopamine (DA) detection, which was prepared by a three-step strategy: the synthesis of Cu-MOF through a solvothermal method, the mixing of Cu-MOF and GO by stirring and sonication, and the reduction of GO by cyclic voltammetry. Jahan et al.29 also reported a complicated work to obtain the satisfactory sensor based on oxygen reduction reaction (ORR). It mainly involved the synthesis of G-dye and linking metalloporphyrin nodes through G-dye to form the hybrid Fe-MOF–GN. The experiment conditions were harsh and the cumbersome process might bring some potential drawbacks such as the structure collapse of MOF due to long time of ultrasonication and high reduction potential of GO. So it is urgent to develop a simple and efficient method to fabricate MOF–GN composites. To the best of our knowledge, one-step synthesis of MOF–GN composites, without intermediate separation steps, has not been reported to date.
In this work, we present a facile one-step method for synthesizing Cu-MOF–GN nanocomposites for the first time. In this method, dimethylformamide (DMF) was used as endogenous reducing agent to reduce GO and as solvent in the synthesis of Cu-MOF. The obtained Cu-MOF–GN nanocomposites were stable and electric conductive. Taking full advantage of the Cu-MOF–GN nanocomposites we constructed H2O2 and ascorbic acid (AA) sensors. The sensors exhibited sufficiently good performance. This indicates that the as-prepared Cu-MOF–GN nanocomposites have promising applications in electrochemical field.
2. Experimental
2.1. Reagents and materials
Cu(NO3)2·3H2O, Na2HPO4·12H2O, NaH2PO4·2H2O, H2O2, DMF, AA and ethanol were purchased from Sinopharm Chemical Reagent Co. Ltd. (Shanghai, China); GO was obtained from Xianfeng Reagent Co. Ltd. (Nanjing, China); 1,3,5-benzene-tricarboxylic acid (H3BTC) was purchased from Aladdin Chemistry Co., Ltd. (Shanghai, China). The support electrolyte was 0.1 M phosphate buffer solution (PBS, pH = 7.0), which was prepared with Na2HPO4·12H2O and NaH2PO4·2H2O.2.2. Apparatus
Cyclic voltammetry and chronoamperometric experiments were performed with a CHI 832C electrochemical workstation (CH Instrument Company, Shanghai, China). A conventional three-electrode system was employed. The working electrode was a modified glassy carbon electrode (diameter: 3 mm), and the auxiliary and reference electrodes were a platinum wire and a saturated calomel electrode (SCE), respectively. The scanning electron microscope (SEM) images were obtained using field emission SEM (ZEISS, Germany). Transmission electron microscopy (TEM) image was obtained by using a JEM-2100 transmission electron microscope with an accelerating voltage of 200 kV. X-ray diffraction data (XRD) were recorded with a PANalytical X'Pert Pro diffractometer (Holland) using Cu Kα radiation (40 kV, 40 mA) with a Ni filter. The Fourier transform infrared (FT-IR) absorption spectra were recorded with a model Nexus-670 spectrometer (Nicolet, USA). Energy dispersive X-ray spectroscopy (EDX) was performed using a Hitachi X-650 SEM (Hitachi Co., Japan).2.3. Synthesis of Cu-MOF, Cu-MOF–GN and GN
A 0.1205 g (0.50 mmol) Cu(NO3)2·3H2O was dissolved in 20 mL DMF and 0.1042 g (0.50 mmol) H3BTC was dissolved in 15 mL DMF. Then they were mixed in a 50 mL round-bottom flask. The mixture was treated by refluxing at 153 °C for 12 h. The obtained blue powder was separated by centrifugation and washed with ethanol and then dried at 80 °C under vacuum.The Cu-MOF–GN nanocomposite was synthesized by dispersing GO powder in the well-dissolved Cu(NO3)2/H3BTC mixture. The resulting suspension was subsequently subjected to the same synthesis procedure as that for Cu-MOF. The amount of GO was changed to make it consisted of 60, 70, 80 or 90 wt% of the final material. As the GO was transferred to GN after the reaction, the resulting composites were denoted as Cu-MOF–GN-1, Cu-MOF–GN-2, Cu-MOF–GN-3, and Cu-MOF–GN-4, respectively.
The GN was prepared by the same procedure for Cu-MOF–GN except not adding Cu(NO3)2·3H2O and H3BTC.35
2.4. Preparation of sensors
Prior to modification, the bare GCE was polished with slurry alumina (1.0, 0.3 and 0.05 μm, respectively), and then washed thoroughly with ultra-pure water with the aid of ultrasonication. To prepare the modified electrodes, 5 mg of the as-obtained sample was dispersed into 1 mL DMF to give homogeneous suspension. Then 4 μL suspension was dip-coated onto the cleaned GCE and the electrode was then dried at room temperature. Thus, the Cu-MOF–GN-1/GCE, Cu-MOF–GN-2/GCE, Cu-MOF–GN-3/GCE Cu-MOF–GN-4/GCE, Cu-MOF/GCE and GN/GCE modified electrodes were obtained, respectively.2.5. Electrochemical measurements
Cyclic voltammograms (CVs) of the sensors were recorded in 0.1 M PBS (pH 7.0) containing 5 mM H2O2 or 3 mM AA. The potential ranged from 0.6 to −0.8 V and −0.3 V to 0.4 V, respectively. The scan rate was 50 mV s−1. For the chronoamperometric measurements the applied potentials were −0.35 V for H2O2 and −0.02 V for AA. The response currents were measured when adding H2O2 or AA under magnetic stirring (Scheme 1). | ||
| Scheme 1 Schematic diagram of the preparation of Cu-MOF–GN and its application to the determination of H2O2 and AA. | ||
3. Results and discussion
3.1. Morphological and structural characterization
To clarify the composite state of Cu-MOF and GN, we performed SEM and TEM measurements. Fig. 1A showed the SEM image of the as-synthesized Cu-MOF particles. The particles were irregular and the size of most crystals was about 1 μm. Fig. 1B displayed the typical wrinkled sheet structure of GN. The GN exhibited excellent dispersibility and stability because DMF could serve as an effective stabilizer of graphene sheets.35,36 Fig. 1C presented the SEM image of Cu-MOF–GN-3. Owing to the cooperative interaction of hydrogen bonding, π–π stacking and Cu–O coordination between Cu-MOF and GN, the Cu-MOF crystals homogeneously grew on the GN host. However, the crystalline order of the Cu-MOF decreased after loading. Moreover, the Cu-MOF crystals agglomerated to some extent and the edges of the crystallites tended to be less sharp as the result of the interference of GN. It is partly due to the restriction effect caused by the carbon skeleton of GN on the growth of Cu-MOF crystallites.24,37 The hybrid of wrinkled graphene layer and the bulk MOF could be observed more clearly through the TEM image in Fig. 1D. | ||
| Fig. 1 SEM images of Cu-MOF (A), GN (B), and Cu-MOF–GN-3 (C). TEM image of Cu-MOF–GN-3 (D). | ||
In order to confirm the full conversion of GO to GN without any additional reductants and the perfect binding between GN and Cu-MOF, power XRD, XPS and FT-IR were carried out further.
As can be seen in Fig. 2A, a feature diffraction peak of GN located at 25° was observed, but no diffraction peak belongs to GO (located at 10°) was detected (curve a), indicating that GO was reduced by DMF thoroughly.38 This characteristic peak and those peaks, which were correspond to (200), (220), (222), (400), (331), (333), (420) and (442) of the octahedral geometry of Cu-MOF (curve b), still remained in Cu-MOF–GN-3 except part overlapping (curve c). Other Cu-MOF–GN-x composites with different mass ratio between Cu-MOF and GN showed similar diffraction peaks (Fig. S1†), and all their XRD patterns were in line with the simulated patterns of Cu-MOF,39 suggesting that the crystallinity of the Cu-MOF had not been disrupted by the incorporation of GN. In addition, the diffraction peaks of GN enhanced gradually with the content of GN increased in the composite. This indicated that Cu-MOF–GN-x nanocomposites were successfully synthesized.
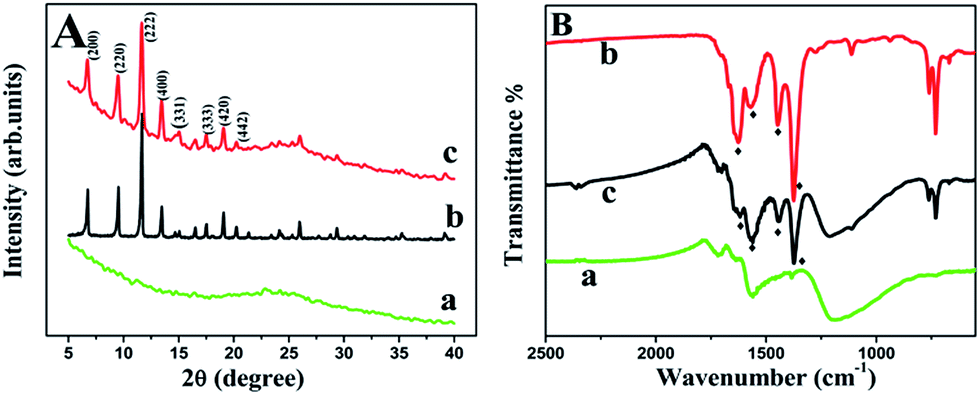 | ||
| Fig. 2 XRD patterns (A) and FT-IR spectra (B) of GN (a), Cu-MOF (b) and Cu-MOF–GN-3 (c). | ||
The reduction level of GN was evaluated further by X-ray photoelectron spectroscopy (XPS) (Fig. S2†). The increased C/O ratio and significantly decreased peaks of C–O and C![[double bond, length as m-dash]](https://www.rsc.org/images/entities/char_e001.gif) O revealed the extinction of oxygen containing groups and the generation of GN. Moreover, the intensity of peak located at 284.4 eV (sp2 carbon) was greatly enhanced compared with that located at 285.3 eV (sp3 carbon), showing the complete reduction from GO to GN.40 It is worth noting that a small N peak centered at 399.5 eV appeared in the XPS of GN (Fig. S2B†), which was supposed to come from N in DMF according to the XPS-peak-differentiating analysis (Fig. S2E†).41 That was due to the adsorption of DMF onto GN.35
O revealed the extinction of oxygen containing groups and the generation of GN. Moreover, the intensity of peak located at 284.4 eV (sp2 carbon) was greatly enhanced compared with that located at 285.3 eV (sp3 carbon), showing the complete reduction from GO to GN.40 It is worth noting that a small N peak centered at 399.5 eV appeared in the XPS of GN (Fig. S2B†), which was supposed to come from N in DMF according to the XPS-peak-differentiating analysis (Fig. S2E†).41 That was due to the adsorption of DMF onto GN.35
The changed FT-IR spectra for before and after hybridization gave more consolidated information about the transformation from GO to GN and the formation of composite. As expected, the absorption peak of CO stretching vibration (at 1725 cm−1) decreased dramatically and the peak of C–O–C (epoxy group) stretching vibration (at 1062 cm−1) disappeared (Fig. S3A†), showing the full reduction. As seen in Fig. 2B, the FT-IR spectrum of Cu-MOF showed four absorption bands at 1626, 1570, 1447, 1375 cm−1, respectively, and Cu-MOF–GN-3 held a rather similar structure to that of Cu-MOF, indicating that the synthesized composites were incorporated with Cu-MOF successfully. The intensity ratio of the absorption peak at 1570 cm−1 to 1626 cm−1 increased with the proportion of GN increasing (Fig. S3B†), which further confirmed that Cu-MOF–GN was synthesized by the proposed one-step method.
In addition, the EDS of Cu-MOF–GN-3 nanocomposite was shown in Fig. S4.† Carbon, oxygen, and copper elements appeared, indicating the existence of Cu-MOF. The nitrogen element was considered to come from the adsorbed DMF on the surface of GN as stated above.
3.2. Thermal stability and electrochemical property of Cu-MOF–GN
As the most concerned properties, thermal stability and electrocatalytic activity have been tested. Herein, the thermal stability of GN, Cu-MOF and Cu-MOF–GN-3 were examined by thermogravimetric analysis (TGA) (Fig. 3A) under a N2 atmosphere. As a result, GN showed less than 4% weight loss near 100 °C, attributable to the removal of water molecules physically adsorbed onto GN. Then the mass loss gradually took place with the increase of temperature. This phenomenon was consistent with the report.35 Cu-MOF began to lose weight below 100 °C due to the elimination of the adsorbed water. Notably, it lost 20 wt% up to 100 °C because of the high adsorbed amount of water by the porous Cu-MOF. From 150 to 230 °C a small weight loss appeared due to the adsorption of DMF. When the temperature was up to 320 °C, a sharp decrease in the weight occurred, suggesting the decomposition of the ligand in Cu-MOF. The TGA curve of Cu-MOF–GN-3 showed the expected thermal behavior. It exhibited about 5 wt% loss up to 100 °C, which was much lower than that of the Cu-MOF because of the decrease of porosity as the incorporation of GN. The sharp weight loss also occurred at 320 °C. These changes were quite consistent with the combined thermal decomposition behaviors of Cu-MOF and GN. This indicated that the composite prepared by this one-step method exhibited equivalent stability with that prepared by other methods.34,42 | ||
| Fig. 3 (A) TGA curves of GN (a), Cu-MOF (b) and Cu-MOF–GN-3 (c); (B) EIS of different modified electrodes in a 0.1 M KCl solution containing 5.0 mM K3Fe(CN)6/K4Fe(CN)6 and from 0.1 Hz to 100.0 kHz; (C) CVs of Cu-MOF–GN-3/GCE in PBS (pH = 7.0) containing 0 (a) and 5 mM (b) H2O2. (D) CVs of different electrodes in 0.1 M PBS (pH = 7.0) with 5 mM H2O2. | ||
Electrochemical impedance spectroscopy (EIS) was used to estimate their electronic conductivity which was closely related to their electrochemical property. As can be seen in Fig. 3B, bare GCE exhibited a semicircle part at high frequency, after being modified with inferior electronic conductivity of Cu-MOF, the diameter of the semicircle significantly increased. On the contrary, the diameter of the semicircle of the GN modified electrode markedly decreased because of its good electronic conductivity. Similarly, the diameter of the semicircle of Cu-MOF–GN-3/GCE was also very small, indicating that Cu-MOF–GN-3 had good conductivity due to GN introducing. The composite could make the electron transfer easier than that of Cu-MOF. The conductivity of Cu-MOF–GN-x changed slightly with composition ratio of GN. The determined charge transfer resistances (Rcts) for Cu-MOF–GN-1, Cu-MOF–GN-2, Cu-MOF–GN-3 and Cu-MOF–GN-4 were 23.6, 18.9, 9.2 and 3.8 Ω respectively. Obviously, the values of Rct reduced with increasing the content of GN, indicating that the electron transfer ability of Cu-MOF has been greatly improved by adding GN.
The electrochemical properties of the Cu-MOF–GN nanocomposites were studied further by cyclic voltammetry with small probe molecules (i.e. H2O2 and AA). As can be seen in Fig. 3C, in blank PBS solutions, the Cu-MOF–GN-3/GCE electrode produced a clear cathodic peak at −0.3 V and a anodic peak at −0.07 V (curve a). A pair of redox peak with similar shape and position had been observed at Cu-MOF/GCE too (Fig. 3D), the redox peak could be considered to come from the redox of copper ion in Cu-MOF.34 After H2O2 was added, the cathodic peak at −0.3 V increased greatly, displaying the good catalytic property of Cu-MOF–GN-3 toward H2O2.
The outstanding electrocatalytic property of Cu-MOF–GN-3/GCE was confirmed further by comparing the response current of H2O2 at different electrodes (Fig. 3D). Obviously, H2O2 did not produce obvious peak at bare GCE. At Cu-MOF/GCE electrode, a small current peak could be seen at about −0.25 V, owning to the redox of copper ion. This process relied on the ingress of electrolyte ions into the Cu-MOF solid lattice through fast internal diffusion.28 Besides, Cu-MOF and GN also exhibited catalytic effect to H2O2. Nevertheless, the most sensitive response of H2O2 was observed at Cu-MOF–GN-3/GCE. This could be ascribed to the good conductivity of Cu-MOF–GN and the synergetically catalytic effect of Cu-MOF and GN.34
Similarly, Cu-MOF–GN-3/GCE showed better electrocatalytic activity to AA, this could be seen from the increased peak current and the negatively shifted peak potential compared with that at GCE (Fig. S5†). The oxidation peak potential of AA at Cu-MOF–GN-3/GCE was around −0.02 V.
3.3. Sensing application
As can be seen in Fig. S6A,† Cu-MOF–GN-3 contained 80 wt% GN was better than others for the electrochemical reduction of H2O2. The peak currents obtained at Cu-MOF–GN-1, Cu-MOF–GN-2, Cu-MOF–GN-3 and Cu-MOF–GN-4 were 226.4, 242.8, 282.3 and 251.3 μA, respectively. The increased current with increasing GN content could be ascribed to the improved conductivity of composites. But increasing the proportion of GN in nanocomposites further would result in decreasing of response current, owning to the weakened electron mediating action of Cu-MOF. So Cu-MOF–GN-3 nanocomposite was selected for preparing modified electrode.
Increasing the volume of Cu-MOF–GN-3 suspension, the peak current of H2O2 increased gradually up to 4.0 μL, then it decreased (Fig. S6B†). This was related to the change of electrode surface area and electron transfer resistance. When the amount of Cu-MOF–GN-3 was too much, the electrode area kept almost unchanged but the resistance increased due to the thickness of Cu-MOF–GN-3 growing. Therefore, 4.0 μL of Cu-MOF–GN-3 suspension was adopted for preparing modified electrode.
The detection potential was optimized too (Fig. S6B†). The response current increased with the operating potential changing from −0.25 V to −0.35 V, but further decreasing the potential, the respond current also decreased. In this case, −0.35 V was chosen.
![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) 180 μM. The linear regression equation was Ip (μA) = 0.05773c (μM) + 15.03 (R2 = 0.995) with a sensitivity of 57.73 μA mM−1. The limit of detection (LOD, S/N = 3) was 2 μM and the current response of Cu-MOF–GN-3/GCE generally reached a steady-state level within 3 s after adding H2O2. Compared with other H2O2 sensors, the Cu-MOF–GN-3/GCE offered wide linear range and good sensitivity (Table S1†).
180 μM. The linear regression equation was Ip (μA) = 0.05773c (μM) + 15.03 (R2 = 0.995) with a sensitivity of 57.73 μA mM−1. The limit of detection (LOD, S/N = 3) was 2 μM and the current response of Cu-MOF–GN-3/GCE generally reached a steady-state level within 3 s after adding H2O2. Compared with other H2O2 sensors, the Cu-MOF–GN-3/GCE offered wide linear range and good sensitivity (Table S1†).
 | ||
| Fig. 4 Amperometric response curves of Cu-MOF–GN-3/GCE upon successive addition of H2O2 at −0.35 V. Left inset: the amperometric response of low concentration of H2O2. Right inset: response time and the relationship between H2O2 concentration and current signal for Cu-MOF–GN-3/GCE. | ||
Simultaneously, under the same conditions for H2O2 except detection potential, the chronoamperometric curves of AA at Cu-MOF–GN-3/GCE were recorded (Fig. 5). The current increased upon the increase of AA concentration, and the linear range was 0.5–6965.5 μM. The linear regression equation was Ip (μA) = 0.01825c (μM) + 2.896 (R2 = 0.993). The limit of detection (LOD, S/N = 3) was 20 nM. Compared with other AA sensors, Cu-MOF–GN-3/GCE offered acceptable performance (Table S2†).
 | ||
| Fig. 5 Amperometric response curves of Cu-MOF–GN-3/GCE upon successive addition of AA at −0.02 V. Left inset: the amperometric response of low concentration of AA. | ||
The repeatability and reproducibility of the developed sensor were evaluated by amperometric determination of 0.1 mM H2O2 and 5 μM AA, respectively. Seven Cu-MOF–GN-3/GCEs were prepared independently by the same way, and the relative standard deviations (RSDs) of the current response were 4.8% (n = 7) (Fig. S7†) and 5.1% (n = 7) for H2O2 and AA, respectively. Seven successive measurements using one electrode gave RSDs of 3.5% (n = 7, for H2O2) and 4.4% (n = 7, for AA). That indicated that the electrode had good reproducibility and repeatability.
The good stability could be confirmed further by comparing the XRD patterns before and after electrochemical measurement. As can be seen in Fig. S8,† the peaks of the resultant were in conformity with those of original Cu-MOF–GN, suggesting that the structure of Cu-MOF–GN-3 had not been damaged during the electrochemical detection. The abundant carbon material (such as GN) seems to act as a strong fixing agent of MOF, which was beneficial to the stability of the crystalline structure of MOF during the electrochemical detection.32,42–44
The storage stability of the sensor was also examined. When the Cu-MOF–GN-3/GCE was stored in a centrifuge tube at 4 °C in a refrigerator for 15 days, the response current for H2O2 retained 96.7% of its initial value, after a month of storage, it still retained 91.5%. These reflected the long-term stability of Cu-MOF–GN-3/GCE.
4. Conclusions
In this study, we have demonstrated for the first time a simple one-step method to synthesis Cu-MOF–GN nanocomposites. The composite was applied to construct sensor for the electrochemical determination of H2O2 and AA. The electrochemical experiments showed that the incorporation of GN greatly enhanced the conductivity, stability and electrocatalysis of CuMOF. The sensor showed excellent electrochemical response, wide linear range and long-term stability. The study would expand the potential applications of MOF-carbon nanocomposite in electrochemical fields.Acknowledgements
This work was supported by the National Natural Science Foundation of China (Grant no. 21075092, 21277105).Notes and references
- M. Eddaoudi, J. Kim, N. Rosi, D. Vodak, J. Wachter, M. O'Keeffe and O. M. Yaghi, Science, 2002, 295, 469–472 CrossRef CAS PubMed.
- K. Xi, S. Cao, X. Peng, C. Ducati, R. V. Kumar and A. K. Cheetham, Chem. Commun., 2013, 49, 2192–2194 RSC.
- J. L. C. Rowsell and O. M. Yaghi, Microporous Mesoporous Mater., 2004, 73, 3–14 CrossRef CAS PubMed.
- P. Horcajada, R. Gref, T. Baati, P. K. Allan, G. Maurin, P. Couvreur, G. Ferey, R. E. Morris and C. Serre, Chem. Rev., 2012, 112, 1232–1268 CrossRef CAS PubMed.
- H. L. Jiang and Q. Xu, Chem. Commun., 2011, 47, 3351–3370 RSC.
- Y. Li and R. T. Yang, Langmuir, 2007, 23, 12937–12944 CrossRef CAS PubMed.
- B. Chen, C. Liang, J. Yang, D. S. Contreras, Y. L. Clancy, E. B. Lobkovsky, O. M. Yaghi and S. Dai, Angew. Chem., Int. Ed., 2006, 118, 1418–1421 CrossRef.
- J. L. Rowsell, E. C. Spencer, J. Eckert, J. A. Howard and O. M. Yaghi, Science, 2005, 309, 1350–1354 CrossRef CAS PubMed.
- J. R. Li, R. J. Kuppler and H. C. Zhou, Chem. Soc. Rev., 2009, 38, 1477–1504 RSC.
- L. Yang, S. Kinoshita, T. Yamada, S. Kanda, H. Kitagawa, M. Tokunaga, T. Ishimoto, T. Ogura, R. Nagumo, A. Miyamoto and M. Koyama, Angew. Chem., Int. Ed., 2010, 122, 5476–5479 CrossRef.
- J. Lei, R. Qian, P. Ling, L. Cui and H. Ju, TrAC, Trends Anal. Chem., 2014, 58, 71–78 CrossRef CAS PubMed.
- M. Jahan, Z. Liu and K. P. Loh, Adv. Funct. Mater., 2013, 23, 5363–5372 CrossRef CAS.
- X. Wang, J. Zhou, H. Fu, W. Li, X. Fan, G. Xin, J. Zheng and X. Li, J. Mater. Chem. A, 2014, 2, 14064 CAS.
- X. Xu, R. Cao, S. Jeong and J. Cho, Nano Lett., 2012, 12, 4988–4991 CrossRef CAS PubMed.
- R. Wu, X. Qian, F. Yu, H. Liu, K. Zhou, J. Wei and Y. Huang, J. Mater. Chem. A, 2013, 1, 11126 CAS.
- J. Mao, L. Yang, P. Yu, X. Wei and L. Mao, Electrochem. Commun., 2012, 19, 29–31 CrossRef CAS PubMed.
- M. Jiang, L. Li, D. Zhu, H. Zhang and X. Zhao, J. Mater. Chem. A, 2014, 2, 5323 CAS.
- P. R. Lima, W. de Jesus Rodrigues Santos, M. O. F. Goulart, A. A. Tanaka, S. M. C. N. Tanaka and L. T. Kubota, Electroanalysis, 2008, 20, 2333–2339 CrossRef CAS.
- Z. Chang, N. Gao, Y. Li and X. He, Anal. Methods, 2012, 4, 4037 RSC.
- L. M. A. Monzon, F. Burke and J. M. D. Coey, J. Phys. Chem. C, 2011, 115, 9182–9192 CAS.
- C. Zhang, M. Wang, L. Liu, X. Yang and X. Xu, Electrochem. Commun., 2013, 33, 131–134 CrossRef CAS PubMed.
- G. Jia, W. Zhang, Z. Jin, W. An, Y. Gao, X. Zhang and J. Liu, Electrochim. Acta, 2014, 144, 1–6 CrossRef CAS PubMed.
- K. F. Babu, M. A. Kulandainathan, I. Katsounaros, L. Rassaei, A. D. Burrows, P. R. Raithby and F. Marken, Electrochem. Commun., 2010, 12, 632–635 CrossRef CAS PubMed.
- Y. Zhang, X. Bo, C. Luhana, H. Wang, M. Li and L. Guo, Chem. Commun., 2013, 49, 6885–6887 RSC.
- H. Hosseini, H. Ahmar, A. Dehghani, A. Bagheri, A. Tadjarodi and A. R. Fakhari, Biosens. Bioelectron., 2013, 42, 426–429 CrossRef CAS PubMed.
- H. Hosseini, H. Ahmar, A. Dehghani, A. Bagheri, A. R. Fakhari and M. M. Amini, Electrochim. Acta, 2013, 88, 301–309 CrossRef CAS PubMed.
- C. Lu, T. Ben, S. Xu and S. Qiu, Angew. Chem., Int. Ed., 2014, 53, 6454–6458 CrossRef CAS PubMed.
- Y. Wang, Y. Wu, J. Xie, H. Ge and X. Hu, Analyst, 2013, 138, 5113–5120 RSC.
- M. Jahan, Q. Bao and K. P. Loh, J. Am. Chem. Soc., 2012, 134, 6707–6713 CrossRef CAS PubMed.
- G. Song, Z. Wang, L. Wang, G. Li, M. Huang and F. Yin, Chin. J. Catal., 2014, 35, 185–195 CrossRef CAS.
- Y. Wang, Y. Wu, J. Xie and X. Hu, Sens. Actuators, B, 2013, 177, 1161–1166 CrossRef CAS PubMed.
- Y. Li, C. Huangfu, H. Du, W. Liu, Y. Li and J. Ye, J. Electroanal. Chem., 2013, 709, 65–69 CrossRef CAS PubMed.
- S. Sharma, G. Kalita, R. Hirano, S. M. Shinde, R. Papon, H. Ohtani and M. Tanemura, Carbon, 2014, 72, 66–73 CrossRef CAS PubMed.
- X. Wang, Q. Wang, Q. Wang, F. Gao, F. Gao, Y. Yang and H. Guo, ACS Appl. Mater. Interfaces, 2014, 6, 11573–11580 CAS.
- K. Ai, Y. Liu, L. Lu, X. Cheng and L. Huo, J. Mater. Chem., 2011, 21, 3365–3370 RSC.
- L. Shang, F. Zhao and B. Zeng, Food Chem., 2014, 151, 53–57 CrossRef CAS PubMed.
- D. Qian, C. Lei, G. P. Hao, W. C. Li and A. H. Lu, ACS Appl. Mater. Interfaces, 2012, 4, 6125–6132 CAS.
- H.-K. Jeong, Y. P. Lee, R. J. W. E. Lahaye, M.-H. Park, K. H. An, I. J. Kim, C.-W. Yang, C. Y. Park, R. S. Ruoff and Y. H. Lee, J. Am. Chem. Soc., 2008, 130, 1362–1366 CrossRef CAS PubMed.
- R. Senthil Kumar, S. Senthil Kumar and M. Anbu Kulandainathan, Electrochem. Commun., 2012, 25, 70–73 CrossRef CAS PubMed.
- X. Li, G. Zhang, X. Bai, X. Sun, X. Wang, E. Wang and H. Dai, Nat. Nanotechnol., 2008, 3, 538–542 CrossRef CAS PubMed.
- S. Delpeux, F. Beguin, R. Benoit, R. Erre, N. Manolova and I. Rashkov, Eur. Polym. J., 1998, 34, 905–915 CrossRef CAS.
- Y. Zhang, A. Nsabimana, L. Zhu, X. Bo, C. Han, M. Li and L. Guo, Talanta, 2014, 129, 55–62 CrossRef CAS PubMed.
- E. Zhou, Y. Zhang, Y. Li and X. He, Electroanalysis, 2014, 26, 2526–2533 CrossRef CAS.
- Y. Zhang, X. Bo, A. Nsabimana, C. Han, M. Li and L. Guo, J. Mater. Chem. A, 2015, 3, 732–738 CAS.
Footnote |
| † Electronic supplementary information (ESI) available: Supplementary results. See DOI: 10.1039/c4ra16950f |
| This journal is © The Royal Society of Chemistry 2015 |