
Morphological and physicochemical properties of Ag–Au binary nanocomposites prepared using different surfactant capped Ag nanoparticles†
Abstract
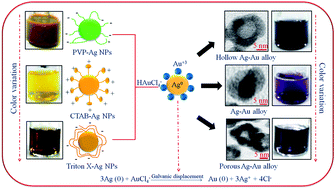
This paper demonstrates the influence of surfactants of different chemical nature passivizing Ag nanoparticles (Ag NPs) on the morphology and physicochemical properties of Ag–Au bimetallic nanostructures. The Ag NPs were synthesized using polyvinylpyrrolidone (PVP), cetyltrimethylammoniumbromide (CTAB) and Triton X-100 (TX), followed by the deposition of Au on their surface. TEM analysis revealed the formation of hollow Ag–Au shells (∼15 nm) and mixed solid Ag–Au alloys (∼20–25 nm) using PVP and CTAB–Ag NPs, respectively, as their reaction templates. In contrast, porous-hollow aggregates of Ag–Au nanostructures (∼16–22 nm) evolved during the reaction between Au3+ and TX–Ag NPs due to the difference in reaction rates between the Au3+ ions and various surfactant capped Ag NPs. As a result, these diverse morphologies of bimetallic nanostructures exhibited a significant variation in surface plasmon (SP) band, color, hydrodynamic size and zeta potential as compared to their monometallic Ag NPs. For example, a SP band of PVP–Ag NPs (488 nm) gradually red-shifted to 550 nm with the addition of Au3+ with notable color change from green to characteristic blue color indicating the composition change from Ag to Au rich. Therefore, the catalytic activity of various Ag–Au bimetallic nanostructures were found to be ∼2 times higher than the monometallic Ag NPs for the reduction of different nitro-aromatic compounds, attributed to the electronic effect at the Ag–Au interface and their morphology.
 Please wait while we load your content...
Please wait while we load your content...

