
An in silico and chemical approach towards small protein production and application in phosphoproteomics†
Ana M. G. C. Diasab,
Olga Iranzo‡
*b and
Ana C. A. Roque *a
*a
aUCIBIO, REQUIMTE, Departamento de Química, Faculdade de Ciências e Tecnologia, Universidade Nova de Lisboa, Campus Caparica, 2829-516 Caparica, Portugal. E-mail: cecilia.roque@fct.unl.pt; Fax: +351 21 294 8550
bAix Marseille Université, Centrale Marseille, CNRS, iSm2 UMR 7313, 13397 Marseille, France. E-mail: olga.iranzo@univ-amu.fr
First published on 9th February 2015
Abstract
The human Pin1 WW domain (hPin1_WW) is a 38 residue protein which specifically recognizes ligands rich in proline and phosphorylated in Ser and Thr residues. This work presents a protocol for the improved chemical synthesis and modification of this protein through automated microwave assisted synthesis combined with the incorporation of pseudoproline units in the protein sequence. After purification, the protein was characterized by Mass Spectrometry and Circular Dichroism spectroscopy with results comparable to the same WW domain chemically synthesized by other strategies or biologically expressed. The protein was further immobilized on a matrix and tested for the selective binding and mild elution of phosphorylated sequences at Ser, Thr and Tyr residues. These results suggest that hPin1_WW is a useful protein scaffold for the purification of phosphorylated species in pTyr and pSer, which can be easily produced and modified by chemical methods.
1. Introduction
Antibodies and derived structures represent the most widespread platform to develop affinity reagents against several targets of medical and biotechnological relevance. Still, the need for stable and robust protein scaffolds amenable of randomization and easy production, has driven several researchers in the quest of alternatives to antibodies.1,2 Such scaffolds include protein domains as for example affibodies (derived from the protein A domain), fibronectin, lipocalins, DARPins and affitins.2,3 WW domains possess 38–40 residues in length, with two conserved tryptophan (W) residues spaced by 20–22 amino acids (which gives the name to the domains family)4 and are assembled on a three β-sheet structure. These motifs bind to proline rich sequences. WW domains are found in at least 200 multidomain proteins and are usually localized in the recognition region known to mediate protein–protein interactions.5 The characteristic structure of the proline residue allows a specific recognition, through weak interactions, which triggers quick responses to the changes in the cellular environment, further translated in signal pathways changes.6 WW domains have been characterized and classified in five different groups based on their recognition sequences.4One of the most widely studied WW domain is found in the human Pin1 protein (hPin1). This protein is a mitotic peptidyl-prolyl isomerase (PPIase) which has been identified as a mitosis cell cycle regulator. Its WW domain is representative of Group IV and specifically recognizes ligands rich in prolines and phosphorylated in Ser and Thr residues.7 Furthermore, hPin1 has been identified in several cellular pathways, since it regulates the isomerisation of phosphorylated residues, and its deregulation has been correlated with cancer, Alzheimer and autoimmune diseases.8 For that reason human Pin1WW (hPin1_WW) domain has been highly studied and its structure is well characterized, as well as its binding mode to phosphorylated peptides.9
Despite their small size, the biological production of WW domains usually requires co-expression with other proteins.10–12 In addition, their chemical synthesis is also challenging because WW domains possess a three β-sheet secondary structure usually regarded as difficult for Solid Phase Peptide Synthesis (SPPS). This is due to the establishment of hydrogen bonds between chains during peptide elongation, resulting in the formation of hydrophobic clusters and peptide aggregation. As a result, the solvation and coupling efficiencies decrease drastically, resulting in truncated peptides and deleted sequences. This turns out in long and difficult purification processes that lower to a large extent the reaction yields.13 However, the chemical synthesis allows the ease incorporation of mutations in the peptide sequence generating variability in the peptides. In addition, when employing automated synthesis, peptides can be obtained in a short period of time. This is a very flexible strategy for peptide production especially useful for peptides shorter than 70 residues, as for higher peptide sizes, the synthesis gets complex, drastically decreasing the product purity and yield.13
Considering the potential of WW domains as scaffold proteins, we optimized the automated microwave assisted SPPS of hPin1_WW through the incorporation of pseudoproline dipeptide units. The stability of the produced structure was analyzed in silico and experimentally. The hPin1_WW was immobilized onto agarose beads to assess the potential of WW scaffolds for the affinity purification of target peptides and proteins.
2. Results and discussion
2.1 In silico studies – stability and affinity of human Pin1 WW domain
The human Pin1 WW domain (hPin1_WWnativeFL, PDB code: 1F8A) was selected as a model WW protein. The amino acid sequences responsible for the folding of the domain and recognition of the targets were maintained,25 but two mutated structures were generated: (i) a smaller native version (hPin1_WWnative) containing only the amino acids important for folding;4 and (ii) a mutated version (hPin1_WWmutated) containing a Cys in the N-terminal (Nt) for subsequent peptide immobilization on a solid support. Additionally, hPin1_WWmutated has a Gly in the C-terminal (Ct) to facilitate the first coupling reaction in SPPS (Scheme 1A). The stability of the hPin1_WW structures was assessed by Molecular Dynamics (MD) simulations (Fig. S1A†). Different parameters were considered, including grid size, and structural features that required adaptations to leave the Cys group outside the grid box – mimicking the immobilization onto a matrix. MD simulations analysis is presented for hPin1_WW in Fig. 1 and S1.† In general, in the Root Mean Square Deviation (RMSD) analysis all the structures of hPin1_WW domain presented similar behavior during the time of simulation. All the structures tend to equilibrate after 20–30 ns and the behavior is retained until the end of the simulation. The Root Mean Square Fluctuation (RMSF) analysis, which informs about the flexibility of each residue in the sequence over the time of the simulation, shows that the flexibility of the smaller versions decreases at the N terminal when compared with the WWnativeFL domain structure. Specifically, for hPin1_WWmutated, there is an increase in rigidity in β-sheet I, Loop II and β-sheet III compared with hPin1_WWnative structure (Fig. 1A). | ||
| Scheme 1 Nomenclature of the sequences of hPin1 WW domain used in this work. (A) Sequences used in the in silico studies derived from PDB code 1F8A file, the mutants were generated using PyMol software. The hPin1_WWnativeFL is the native full length hPin1 WW domain (42 residues); hPin1_WWnative, corresponds to a sequence of 37 residues; and hPin1_WWmutated, corresponds to hPin1_WWnative mutated in the N terminal with a Cys and an introduction of a Gly in the C terminal (see Experimental section for details); (B) strategy for SPPS using pseudoproline units (see Experimental section for details), where X1 = pseudoproline unit Fmoc-Ile-Thr(psiMe, Mepro)-OH and X2 = pseudoproline unit Fmoc-Ser(tBu)-Ser(psiMe, Mepro)-OH; amino acids in italic needed special couplings, and for amino acids in green and underlined, a capping step was added. The structures of the pseudoproline units used are sketched. The chemical structures sketches were designed using Marvin Beans version 6.3.0 (ChemAxon). | ||
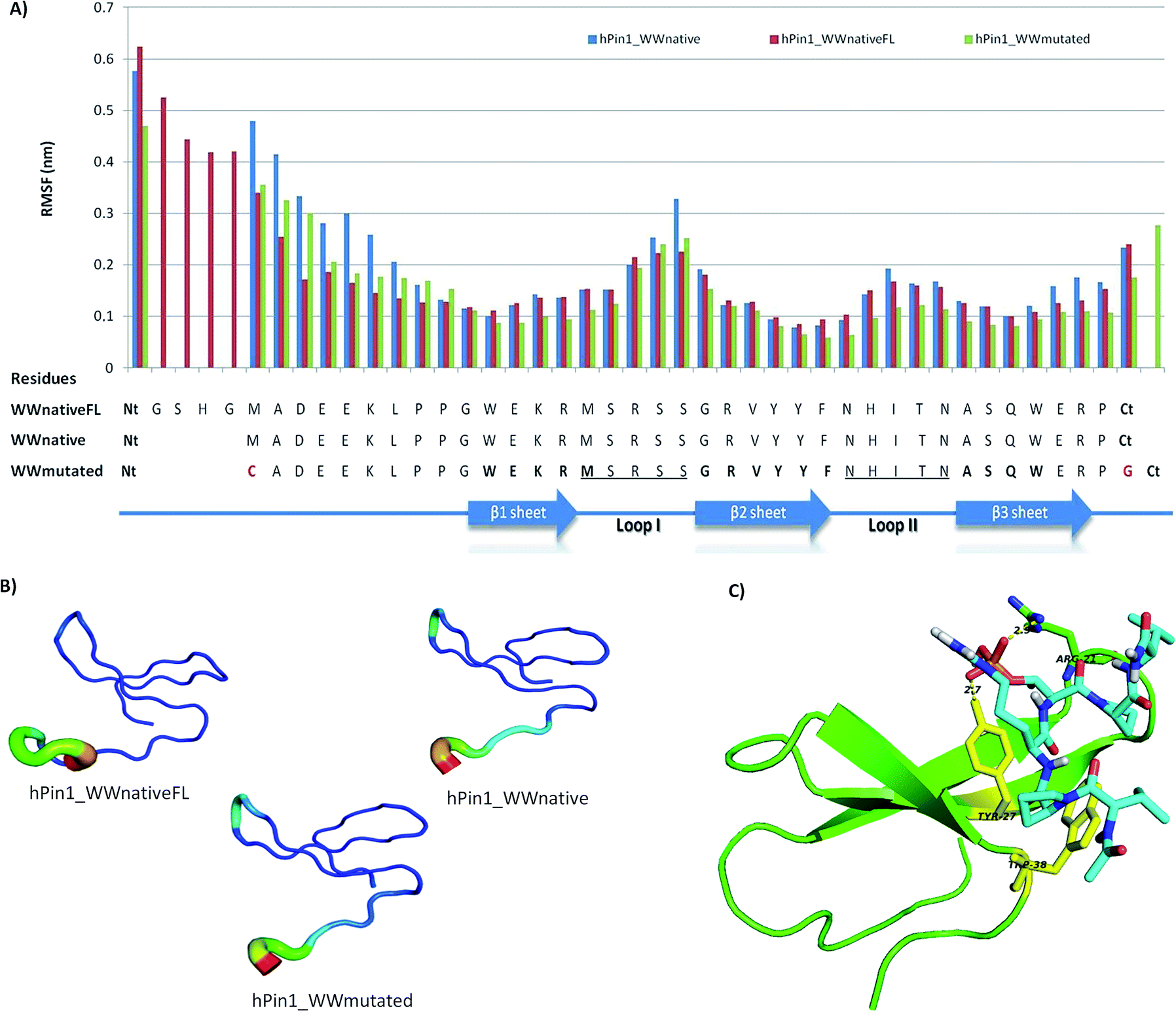 | ||
| Fig. 1 In silico studies results. (A) Root Mean Square Fluctuation (RMSF) that analyses the flexibility of each residue in the structure over the time of simulation (B) B-factor analysis that identifies the most flexible residues during simulation compared with the initial structure that was simulated; the results of the analysis are displayed in a color mode (most flexible residues in red and less flexible in dark blue). (C) Best docking solution between hPin1 and peptide VP*RpTPV. The H-bond interactions between the phosphorylated group in the Thr and Arg-21 (loopI) and Tyr-27 (β-sheet II) are identified. In yellow are represented the residues Tyr-27 and the Trp-38 that are described in the literature as part of the “X-P-groove” where the Pro* is recognized. | ||
The β-sheets from WW domains are less flexible, as they establish hydrogen bond interactions between each other, as observed in the H-bond interactions analysis (Fig. S1B and C†). In particular, the interactions between the backbone of amino acids residues Asn27–Ala32, Gln34–Tyr25, Glu13–Phe26, and Tyr24–Arg15 are important for the characteristic tertiary structure and WW domain stability.11 The most flexible region is Loop I, since it is involved in the molecular recognition of ligands in the hPin1_WWnativeFL structure,4,26 but we still observed H-bond interactions between Ser17–Arg22 that are important for loop maintenance.11,27
For the affinity studies the WW domain was used as a receptor and different peptides as ligands. The ligands tested were the native phosphorylated ligand, the non-phosphorylated version and a proline-rich peptide, and their structures were docked at the surface of WW domain using a blind docking strategy. As the three ligands tested possess proline residues, the recognition of the peptides through the consensus “X-P-groove region” formed by Tyr-27 and Trp-38 residues from WW domain was observed (Fig. S2†). The proline rich peptide presented the highest affinity towards the WW domain (Ka 2.49 × 107 M−1). In this case, apart from the “X-P-groove region” the other proline residues are also recognized through hydrophobic interactions established with other WW domain residues. Regarding the phosphorylated peptide, the best binding conformation and the estimated affinity constant were similar to those described in the literature (Ka 2.28 × 105 M−1 versus Ka 2.04 × 105 M−1 (ref. 9)) (Fig. 1C and Table SI†). For the non-phosphorylated peptide version, the binding conformation is similar to the phosphorylated peptide but there is a decrease in affinity of one order of magnitude, due to the lack of hydrogen bond formation between Arg-21 and the phosphate group. These results can be explained due to the conserved recognition in WW domains of the Pro residue by a Tyr and Trp residues in the β-sheet. This recognition event occurs first, and restricts the liberty of the other residues in the peptide ligand.28 Additionally, in the case of hPin1_WW, when peptides contain pSer/pThr residues the binding affinity increases as the phosphate group interaction with the loop I residues will contribute positively for the binding. From these in silico studies was possible to conclude that the hPin1_WWmutated structure maintains its stability and molecular recognition sites against the native ligands of hPin1_WW.
2.2 Peptide solid-phase chemical synthesis using pseudoproline units
After studying in silico the stability of hPin1_WWmutated, this structure was chemically synthesized by Fmoc-based SPPS in an automated microwave system. Firstly, the hPin1_WWmutated peptide was produced using a standard protocol for SPPS (Table SII†). Using this approach, several co-products were obtained as shown in the HPLC chromatograms (Fig. S3A†) difficulting the purification step and therefore the overall yield of the synthesis.Besides microwave technology, that promotes better coupling reaction between amino acids due to the kinetic power input,13,29 different strategies have been described in the literature to overcome problems with difficult chemical synthesis. These include resins with an hydrophobic character (e.g. poli(ethyleneglicol) (PEG) based resins);30 different solvent mixtures;31 chaotropic salts;13 pseudoproline units which are oxazolidine dipeptides known to disrupt secondary structure;32 and fragments condensation using native chemical ligation.33,34
We explored the use of a PEG based resin and the incorporation of pseudoproline dipeptide units during synthesis.32,35 This was possible because hPin1_WWmutated domain possess Ser and Thr residues, and their location in the sequence is appropriate for the insertion of pseudoproline units32 (see Scheme 1B for details). Additionally, we introduced capping steps after the 23rd amino acid. The synthesized peptide (hPin1_WWmutated) was purified by reverse-phase preparative HPLC (Fig. S3B†), and its purity determined by reverse-phase analytical HPLC (Fig. 2C). ESI-MS analysis (Fig. S4†) confirmed the identity of the peptide. For hPin1_WWmutated, the production yield was 30% and the purity 95%. The hPin1_WWmutated was obtained through SPPS in less than 24 h and this strategy was repeated four times with high reproducibility. In each production cycle, 150–200 mg of peptide was obtained which demonstrates the robustness of the methodology.
 | ||
| Fig. 2 Circular dichroism studies: (A) far-UV CD spectra of hPin1_WWmutated (50 μM) at 2 °C and 98 °C in 20 mM MOPS pH 7.2, 100 mM NaCl; (B) fitting to the two state-model curve for determination of melting temperature (Tm); (C) far-UV CD spectra obtained at 4 °C using different buffers: 10 mM HEPES buffer pH 7, 10 mM sodium acetate pH 4, and 10 mM CAPS buffer pH 10 (all with 100 mM NaCl) and the regeneration buffer used for the solid support: 0.1 M NaOH in 30% isopropanol. | ||
Our yield results are difficult to compare with the literature since few authors report the actual yields obtained during chemical and biological production of WW domains. A similar yield (∼25%) was reported for the manual synthesis of another WW domain with 37 residues, FBP28 (Formin-binding protein), using also pseudoproline units, but with considerable longer synthesis time (∼60 h versus ∼24 h in this work).32 These data highlights the benefits of using microwave technology (efficient kinetic power input) combined with automated systems.35,13
2.3 Peptide characterization through circular dichroism spectroscopy
Circular Dichroism (CD) spectroscopic studies were performed to determine the secondary structure and stability of the chemically synthesized hPin1_WWmutated. The spectra were recorded in the Far-UV region to check for the characteristic signals of the WW domains. These proteins have a three β-sheet structure showing a CD spectrum with a maximum positive ellipticity at ∼230 nm and a maximum negative ellipticity at 206 nm.12 Fig. 2A shows the CD spectra obtained for hPin1_WWmutated at 2 °C in MOPS buffer pH 7.2, which shows a maximum positive ellipticity at 226 nm, the specific signal for hPin1_WW.23 In addition, temperature denaturation experiments were performed to determine peptide stability. The change in ellipticity at 230 nm was monitored as temperature increased from 2 °C to 98 °C (Fig. 2B). The CD unfolding curve was fitted to a two state-model to determine the Tm of hPin1_WWmutated, using the equation described by Koepf et al.12 The Tm determined using these conditions was 56.3 ± 0.5 °C (R2 = 0.998) (Fig. 2B). This result is in agreement with the Tm value of 55 °C determined by Kraemer-Pecore et al. for hPin1_WW (34 residues) under the same buffer conditions.23 Different Tm values have been reported for native hPin1_WW at pH 7 using different buffer conditions. Kaul et al.36 reported a Tm of 58 °C (36 residues length, 10 mM sodium phosphate buffer) and Deechongkit et al.37 reported Tm values of 59 °C (34 residues length, 20 mM sodium phosphate buffer, 1 mM DTT). Interestingly, the same authors observed a 7 °C decrease in Tm values when the same structure was capped in the N and C-terminal with acetyl and amide group, respectively. This information suggests that the charges in the terminal regions of the peptide provide stability to the overall structure.37hPin1_WWmutated will be immobilized onto a solid support to explore its capability to capture target peptides and proteins. For that purpose different buffer conditions were tested (10 mM HEPES buffer pH 7; 10 mM sodium acetate buffer pH 4; 10 mM CAPS buffer pH 10 with 100 mM NaCl; and finally, a regeneration buffer: 0.1 M NaOH in 30% isopropanol) and their influence on the structure of the hPin1_WWmutated analyzed by CD spectroscopy. The results presented in Fig. 3C indicate that the signal at 226 nm of hPin1_WWmutated is maintained despite the nature and pH of the buffer. When temperature was increased to 23 °C, no major differences in the signal were observed.
 | ||
| Fig. 3 Binding studies at different pH values for phosphorylated and non-phosphorylated peptides in batch mode using 100 mg of hPin1WW_Ag. (A) Binding percentage and (B) peptide bound mass/support mass. VV, non-phosphorylated native peptide – VPRTPV; VVP, phosphorylated native peptide – VPRpTPV; PP, pro-rich peptide – PPPPYPAW; VV-B, agarose unmodified tested with VV; VVP-B, agarose unmodified tested with VVP and PP-B, agarose unmodified tested with PP. Binding percentage is calculated using (mass peptide loaded – mass peptide output) × 100/mass peptide loaded and peptide bound mass/support mass (μg mg−1) is calculated using (mass peptide loaded − mass peptide output)/mass support, (number of repeats n = 2). | ||
2.4 Immobilization of hPin1_WWmutated onto solid support for affinity purification
Phosphorylation is one of the most important post translational modification in proteins, and it has been implicated in several steps in cellular regulation.38 As this modification can be temporary and reversible in proteins, the recognition of phosphorylated and non-phosphorylated proteins can work as a molecular switch. Kinases and phosphatases are responsible for phosphorylation state modification. For that reason, perturbations in the equilibrium of the natural protein modifications have been correlated with several diseases.8 These observations have increased the interest in the phosphoproteome, which relates all the knowledge obtained about these modifications through different methods: phospho-enrichment methods and mass spectrometry (MS).39 Since hPin1_WW domain it is a natural binding motif of phosphorylated peptides, we aim to immobilize it in solid support to demonstrate its application in the enrichment/purification of phosphorylated species.For the immobilization of the hPin1_WWmutated domain, the chemistry of sulfo-SMCC (sulfosuccinimidyl-4-[N-maleimidomethyl]cyclohexane-1-carboxylate) was chosen using cross-linked agarose as solid-support (hPin1_WWAg). The immobilization yield was 60% (2,45 × 10−3 μmol peptide per mg support) and the strategy was very reproducible. After immobilization the adsorbent hPin1_WWAg was tested with pure peptide solutions for binding, namely the native ligand peptide (VPRpTPV, VVP), the non-phosphorylated version (VPRTPV, VV), and also the proline-rich peptide (PPPPYPAW, PP). Since hPin1_WW is a natural phosphate binding domain, we aimed first to maximize the conditions to recognize the phosphate group, which possesses different charges at different pHs. To determine the best pH value we tested three different buffers and conditions: pH 4 (–OPO3H−1); pH 7 and pH 10 (–OPO3H−1/−2),40 the buffers were chosen to avoid phosphate groups which act as competitors. The buffers selected were 25 mM sodium acetate buffer at pH 4; 25 mM HEPES buffer at pH 7; 25 mM CAPS buffer at pH 10, with 150 mM NaCl.
The results for a small scale of adsorbent (100 mg) are shown in Fig. 3, where agarose with no modification was used as a control. At pH4 the peptides presented very low binding values, as at this buffer condition the support is positively charged and the peptides too, so there is low electrostatic attraction, i.e. low binding. The exception is observed for VVP that has a negative charge (–OPO3H−1) and for that reason can present some binding to the unmodified support. At pH10, hPin1_WWAg is negatively charged (−2) and peptide VV is positively charged, this will promote the electrostatic attraction to the support and high binding. However, the peptide VVP presents a phosphate group (negative charge) and thus, its overall positive charge is decreased which results in lower electrostatic attractions, i.e. low binding. The same is observed for the PP peptide that at this pH is negatively charged. Considering the unmodified support, agarose at pH 10 presents a negative charge and consequently the VV will present the highest binding as opposed to the other tested peptides.
The small differences observed between Fig. 3A and B are due to the fact that in Fig. 3A the data is normalized by the mass of peptide loaded whereas in Fig. 3B it is normalized by the mass of support used, as there are slight differences between each assay.
From these results, we selected the buffer condition where the phosphorylated peptide had the highest binding and the non-phosphorylated version had the lowest binding, thus, HEPES buffer at pH 7. Under this condition, the proline-rich peptide was also recognized by the support, however in less extent than the native phosphorylated ligand. The binding percentage for the VPRpTPV is 30% (0.14 μg peptide bound per mg support). We repeated the screening of the phosphorylated peptide with 3× the solid support mass (300 mg), and the binding percentage increased for 85% (0.16 μg peptide bound per mg support).
After this study, the affinity of hPin1_WWAg was tested for a model protein, Bovine Serum Albumin (BSA) chemically modified with phosphate groups: phosphothreonine (BSA-pT), phosphoserine (BSA-pS) and phosphotyrosine (BSA-pY), which are the most frequent modifications observed in proteins. As a control, we tested unmodified BSA (Fig. 4A and B). The binding percentage was higher for the BSA-pS and BSA-pY than for BSA-pT and ranged from 35% (0.059 μg protein bound per mg support) to 40% (0.065 μg protein bound per mg support). The binding for the BSA-pT and unmodified BSA was approximately 5%. These results indicate that the recognition is made through the phosphorylated group, but also by interactions with other residues in the protein that contribute for the binding. One elution condition was tested for the BSA-pS and BSA-pY, using a competitive strategy with phosphate groups and mild conditions (PBS buffer). The elution was successful for BSA-pS (0.054 μg protein eluted per mg support) and BSA-pY (0.072 μg protein eluted per mg support), which corresponds to protein recoveries of 92% and 90% respectively for BSA-pS and BSA-pY (Fig. 4A and B).
 | ||
| Fig. 4 (A) Binding studies using 25 mM HEPES buffer + 150 mM NaCl at pH 7 for proteins BSA-pT, BSA-pS, BSA-pY and BSA and (B) elution with 50 mM Phosphate buffer + 50 mM NaCl, pH 7. (n.d. – non determined). Elution percentages are calculated as (mass protein eluted) × 100/mass protein loaded (number of repeats n = 2). | ||
3. Conclusions
The synthetic approach presented here shows the production of the hPin1_WW domain with the characteristic folding of a WW domain and a Tm value in agreement with those reported in the literature. The pseudoproline units strategy using automated microwave technology demonstrated to be a robust, less time consuming and an extremely efficient route for the production and modification of this small protein with good yield.It was also shown that hPin1_WWmutated immobilized in a solid support maintains the affinity for the native phosphorylated ligand VPRpTPV. From the binding studies with BSA and their modified versions with pSer and pTyr, we conclude that it has affinity for these modified proteins and thus recognition is mediated by the phosphorylated residues. In addition, it is possible to completely recover the protein bound to the solid support under mild conditions (competitive elution with phosphate buffer at neutral pH). In summary, the data presented here show that the chemical production of hPin1_WW at high yields is possible as well as its immobilization onto solid supports. Therefore, we demonstrate a novel platform for the chemical production and modification of a small protein scaffold and its application for the purification of phosphorylated proteins and peptides.
4. Experimental
4.1 In silico studies
4.2 Docking studies – affinity studies
The affinity studies were conducted between hPin1_WWmutated structure (receptor) and several peptides (ligands): VPRpTPV (native ligands of hPin1_WWnative domain),9 the non-phosphorylated version of the same peptide and PPPPYP (a proline-rich peptide derived from the natural ligand of YAP65 WW domain).12 The peptides were designed using Pymol 1.3 software and saved in a pdb file format. The coordinates of receptor and ligand were converted into pdbqt file format. The grid parameter files were setup using the AutoDock 4.0.7 tool package.21 A Cubic grid was defined using grid size [100 100 80]; spacing of 0.375 Å and a Grid center coordinates of [−5.826 8.505 54.406]. Since the aim of the work was to immobilize hPin1_WW into a solid support, the box was defined such that the Cys residue was maintained out of the box. The ligands had degree of freedom and were allowed to explore the surface of the receptor. The Lamarckian genetic algorithm was used and a maximum number of 256 solutions were tested. In the solutions analysis was requested that the structures were grouped with a RMS tolerance of 4 Å. After docking, the solutions were analyzed based on an energy criteria – best scored clusters, with lower estimated binding free energy, and also based on number of elements criteria – from the best scored clusters only the ones with highest number of elements were further analyzed. Thus we aimed to select the best binding conformation, based in the energy score and the frequency of this conformation. The results were visualized using PyMol 1.3 software.4.3 Peptide chemical synthesis and characterization
3-(N-Morpholino)propanesulfonic acid (MOPS); 4-(2-hydroxyethyl)piperazine-1-ethanesulfonic acid (HEPES); 3-(cyclohexylamino)-1-propane sulfonic acid (CAPS), ethylenediaminetetraacetic acid (EDTA) where used in buffers preparation and were obtained from Sigma-Aldrich (Loures, Portugal).
The reagents used in these procedures were always high grade.
The first synthesis of hPin1_WWmutated was carried out in a CEM Liberty microwave peptide synthesizer (CEM Corporation, North Carolina, USA) using the standard protocol provided by the manufacturer (Table SII†). A Rink amide MBHA resin 100–200 mesh with a substitution of 0.59 mmol g−1 was employed (scale 0.25 mmol). All the amino acid reaction couplings were undertaken at 75 °C, except for Asp, His and Cys that were carried out at 50 °C. The deprotection and cleavage of the peptide was carried out using the standard protocol, namely TFA/thioanisole/1,2-ethanedithiol/anisole (%v/v = 90![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) :5:3:2) for 2 h at room temperature. The resin was filtered out and the TFA solution was reduced under a stream of nitrogen. Cold diethyl ether was added to this solution to precipitate the peptide, which was then filtered and washed again with diethyl ether. The crude peptide was dissolved in water and lyophilized. The reverse-phase HPLC chromatogram of the crude peptide showed a mixture of products (Fig. S3A†).
:5:3:2) for 2 h at room temperature. The resin was filtered out and the TFA solution was reduced under a stream of nitrogen. Cold diethyl ether was added to this solution to precipitate the peptide, which was then filtered and washed again with diethyl ether. The crude peptide was dissolved in water and lyophilized. The reverse-phase HPLC chromatogram of the crude peptide showed a mixture of products (Fig. S3A†).
Pseudoproline units were used for the second synthesis of hPin1_WWmutated. This strategy was feasible because this domain contains Ser and Thr residues in the middle of the sequence (Scheme 1B). Consequently, two dipeptide units, Fmoc-Ser(tBu)-Ser(psiMe, Mepro)-OH and Fmoc-Ile-Thr(psiMe, Mepro)-OH were introduced during synthesis (see Scheme 1B). hPin1_WWmutated was assembled in a NovaPEG Rink amide resin (substitution 0.64 mmol g−1) previously swelled for 30 min in DMF and directly loaded in the reaction vessel. All the amino acid reaction couplings were undertaken at 75 °C, except for Asp, His and Cys that were carried out at 50 °C (Table SII†). Double coupling was used for the pseudoproline units, the amino acids following these units and the amino acids Arg, Pro, Trp, Thr and Val. After the loading of the 23rd amino acid, a capping reaction step was introduced following each amino acid coupling to prevent the formation of truncated sequences and the coupling and deprotection reaction times were slightly increased. The peptide was acetylated at the N-terminal. Cleavage and removal of the protecting groups was simultaneously performed by treatment with the mixture TFA/thioanisole/1,2-ethanedithiol/anisole (%v/v = 90:5:3:2) for 3 hours at room temperature and under nitrogen. The solution was reduced under a nitrogen stream and cold diethyl ether was added to precipitate the crude peptide which was dissolved in water and lyophilized.
hPin1_WWmutated was purified by preparative reverse-phase HPLC (Phenomenex Jupiter Proteo column, 250 mm × 21.2 mm, 4 μ, 90 Å) using solvent A (water/TFA, 99.9:0.1 v/v) and solvent B (acetonitrile/water/TFA, 90:9.9:0.1 v/v). The peptide produced using the standard method was eluted with a linear gradient of solvent B (10% to 50% in 25 min) at a flow rate of 10 mL min−1 (Fig. S3 A,† Rt = 18.8 min). The peptide produced using pseudoproline units was eluted with a linear gradient of solvent B (20% to 44% in 20 min) at a flow rate of 10 mL min−1 (Fig. S3B,† Rt = 15.8 min). The purity of the peptide was checked by analytical reverse-phase HPLC (Phenomenex Jupiter Proteo column, 250 mm × 4.6 mm, 4 μ, 90 Å) using a linear gradient of solvent B (10–50% in 20 min) at a flow rate of 0.6 mL min−1 (Fig. S3C,† Rt = 18.2 min) and it was greater than 95%. The pure hPin1_WWmutated was characterized by ESI-MS (Fig. S4†).
where θobs is the observed ellipticity in millidegrees, C is the molar concentration, l is the path length of the cell in cm, residue is the number of residues in the sequence and 10 is a conversion factor to dmol. Tm values were determined using the protocol described by Kraemer-Pecore et al.23

4.4 Immobilization of hPin1_WWmutated and affinity studies
To proceed with immobilization of hPin1_WWmutated in the aminated support, the following solutions were used: the immobilization buffer, 50 mM HEPES, 150 mM NaCl, 1 mM EDTA pH 7.2 and to dissolve sulfosuccinimidyl 4-[N-maleimidomethyl]cyclohexane-1-carboxylate (Sulfo-SMCC), a 50 mM HEPES pH 7 buffer. These buffers were de-aerated using a nitrogen stream before use. Sulfo-SMCC was used in an excess of 5× with ratio of 0.5 μmol peptide: 1 μmol of aminated support. The support was washed with 5 × 500 μL immobilization buffer. The solution of Sulfo-SMCC was prepared and added to the support and incubated with rotative agitation at 30 rpm for 30 min at room temperature (23 °C). During this time, a solution of hPin1_WWmutated (10 mg mL−1) was prepared in the immobilization buffer and sonicated for 5 min to facilitate the dissolution. To avoid any oxidized Cys in the peptide, Bond-Breaker TCEP Solution (Pierce, ThermoScientific) in a ratio of 1:100 was added. After incubation the solution was removed by filtration and the support was washed 3× with aerated buffer to eliminated unreacted Sulfo-SMCC. After this, the solution of hPin1_WWmutated was added to the activated support and incubated for 1 h. The support was washed with immobilization buffer for 4× to remove unreacted peptide. To block the unreacted sites in the support, a solution of l-cystein (prepared in the same proportion as Sulfo-SMCC solution) was incubated with the support for 30 min. Following, the support was washed 4×. Finally, to determine the amount of hPin1_WWmutated that was immobilized in the support, the solution of hPin1_WWmutated that was loaded and all the washes after immobilization were quantified by Abs280 nm. The support (hPin1_WWAg) was saved at 4 °C in immobilization buffer.
The peptides VPRpTPV and VPRTPV (purity >95% from GeneCust-Europe), and PPPPYPAW (purity >97% from CASLO, ApS, Lyngby, Denmark) with no modifications in the N and C terminal (free terminals). Peptides solutions were added in 50 μg to 100 mg hPin1_WWAg and the studies were performed using solutions of 300 μL. The proteins Bovine Serum Albumin (BSA), phosphothreonine-BSA (BSA-pT), phosphoserine-BSA (BSA-pS) and phosphotyrosine-BSA (BSA-pY) were from Sigma-Aldrich (Germany). Solutions of these proteins were prepared with a mass range of 50 μg in a 500 μL total volume. These solutions were incubated with 300 mg of hPin1_WWAg. Each peptide and protein was tested individually with the hPin1_WWAg. The flow-through, 5× washes and 5× elution samples were analyzed at Abs230 nm for the VPRpTPV and VPRTPV and at Abs280 nm for PPPPYPAW and BSA proteins. The Binding Percentage was calculated using (mass peptide loaded − mass peptide output) × 100/mass peptide loaded. The peptide bound mass/support mass (μg mg−1) was calculated using (mass peptide loaded − mass peptide output)/mass support, elution percentages for proteins were calculated as (mass protein eluted) × 100/mass protein loaded.
Acknowledgements
A.M.G.C.D. would like to thank Dr Iris Batalha and Dr Ricardo Branco for the helpful discussions and technical support in the immobilization and in the in silico studies, respectively. A.M.G.C.D. is grateful to Fundação para a Ciência e Tecnologia (FCT) – Portugal for funding through PhD grant SFRH/BD/72664/2010. This work is funded by National Funds through FCT under the project PEst-C/EQB/LA0006/2011, Pest-OE/EQB/LA0004/2011, the National Network of Mass Spectrometry REDE/1504/REM/2005, project PTDC/EBB-BIO/102163/2008, PTDC/EBB-BIO/118317/2010 and ERA-IB-2/0001/2013. Mass Spectrometry data were provided by the Mass Spectrometry Laboratory, Analytical Services Unit, Instituto de Tecnologia Química e Biológica, Universidade Nova de Lisboa.References
- V. Marx, Nat. Methods, 2013, 10, 829–833 CrossRef CAS.
- H. K. Binz, P. Amstutz and A. Plückthun, Nat. Biotechnol., 2005, 23, 1257–1268 CrossRef CAS PubMed.
- N. Sawyer, E. B. Speltz and L. Regan, Biochem. Soc. Trans., 2013, 41, 1131–1136 CrossRef CAS PubMed.
- M. J. Macias, V. Gervais, C. Civera and H. Oschkinat, Nat. Struct. Biol., 2000, 7, 375–379 CrossRef CAS PubMed.
- M. Sudol and T. Hunter, Cell, 2000, 103, 1001–1004 CrossRef CAS PubMed.
- A. Zarrinpar, R. P. Bhattacharyya and W. A. Lim, The Structure and Function of Proline Recognition Domains, 2003 Search PubMed.
- P. J. Lu, X. Z. Zhou, M. Shen and K. P. Lu, Science, 1999, 283, 1325–1328 CrossRef CAS PubMed.
- Y.-C. Liou, X. Zhen Zhou and K. Ping Lu, Trends Biochem. Sci., 2011, 36, 501–514 CrossRef CAS PubMed.
- M. A. Verdecia, M. E. Bowman, K. P. Lu, T. Hunter and J. P. Noel, Nat. Struct. Mol. Biol., 2000, 7, 639–643 CAS.
- H. Yanagida, T. Matsuura, Y. Kazuta and T. Yomo, ChemBioChem, 2011, 12, 962–969 CrossRef CAS PubMed.
- M. Jäger, H. Nguyen, J. C. Crane, J. W. Kelly and M. Gruebele, J. Mol. Biol., 2001, 311, 373–393 CrossRef PubMed.
- E. K. Koepf, H. M. Petrassi, M. Sudol and J. W. Kelly, Protein Sci., 1999, 8, 841–853 CrossRef CAS PubMed.
- S. L. Pedersen, A. P. Tofteng, L. Malik and K. J. Jensen, Chem. Soc. Rev., 2012, 41, 1826–1844 RSC.
- B. Hess, C. Kutzner, D. v. d. Spoel and E. Lindahl, J. Chem. Theory Comput., 2008, 4, 435–447 CrossRef CAS.
- X. Daura, A. E. Mark and W. F. V. Gunsteren, J. Comput. Chem., 1998, 19, 535–547 CrossRef CAS.
- H. J. C. Berendsen, J. P. M. Postma, W. F. Van Gunsteren, A. DiNola and J. R. Haak, J. Chem. Phys., 1984, 81, 3684–3690 CrossRef CAS.
- G. Bussi, D. Donadio and M. Parrinello, J. Chem. Phys., 2007, 126, 1–7 CrossRef PubMed.
- B. Hess, H. Bekker, H. J. C. Berendsen and J. G. E. M. Fraaije, J. Comput. Chem., 1997, 18, 1463–1472 CrossRef CAS.
- W. L. DeLano, The PyMOL Molecular Graphics System, DeLano Scientific LLC, San Carlos, CA, USA, 2002 Search PubMed.
- W. Humphrey, A. Dalke and K. Schulten, J. Mol. Graphics, 1996, 14, 33–38 CrossRef CAS PubMed.
- G. M. Morris, D. S. Goodsell, R. S. Halliday, R. Huey, W. E. Hart, R. K. Belew and A. J. Oslon, J. Comput. Chem., 1998, 19, 1639–1662 CrossRef CAS.
- Protein Structure: a Practical approach, ed. T. E. Creighton, IRL Press at Oxford University, Oxford, 2nd edn, 1997 Search PubMed.
- C. M. Kraemer-Pecore, J. T. J. Lecomte and J. R. Desjarlais, Protein Sci., 2003, 12, 2194–2205 CrossRef CAS PubMed.
- J. M. Haigh, A. Hussain, M. L. Mimmack and C. R. Lowe, J. Chromatogr. B: Anal. Technol. Biomed. Life Sci., 2009, 877, 1440–1452 CrossRef CAS PubMed.
- H. I. Chen, A. Einbond, S.-J. Kwak, H. Linn, E. Koepf, S. Peterson, J. W. Kelly and M. Sudol, J. Biol. Chem., 1997, 272, 17070–17077 CrossRef CAS PubMed.
- T. Sharpe, A. L. Jonsson, T. J. Rutherford, V. Daggett and A. R. Fersht, Protein Sci., 2007, 16, 2233–2239 CrossRef CAS PubMed.
- T. Peng, J. S. Zintsmaster, A. T. Namanja and J. W. Peng, Nat. Struct. Mol. Biol., 2007, 14, 325–331 CAS.
- C.-e. A. Chang and Y.-m. M. Huang, in Annual Reports in Computational Chemistry, ed. A. W. Ralph, Elsevier, 2013, vol. 9, pp. 61–84 Search PubMed.
- B. Bacsa, K. Horvati, S. Bosze, F. Andreae and C. O. Kappe, J. Org. Chem., 2008, 73, 7532–7542 CrossRef CAS PubMed.
- B. de la Torre, A. Jakab and D. Andreu, Int. J. Pept. Res. Ther., 2007, 13, 265–270 CrossRef CAS.
- A. K. Tickler, A. B. Clippingdale and J. D. Wade, Protein Pept. Lett., 2004, 11, 377–384 CrossRef CAS PubMed.
- I. Coin, M. Beyermann and M. Bienert, Nat. Protoc., 2007, 2, 3247–3256 CrossRef CAS PubMed.
- Z. Fidan, A. Younis, P. Schmieder and R. Volkmer, J. Pept. Sci., 2011, 17, 644–649 CrossRef CAS PubMed.
- P. Siman, S. V. Karthikeyan and A. Brik, Org. Lett., 2012, 14, 1520–1523 CrossRef CAS PubMed.
- P. Marek, A. M. Woys, K. Sutton, M. T. Zanni and D. P. Raleigh, Org. Lett., 2010, 12, 4848–4851 CrossRef CAS PubMed.
- R. Kaul, A. R. Angeles, M. Jäger, E. T. Powers and J. W. Kelly, J. Am. Chem. Soc., 2001, 123, 5206–5212 CrossRef CAS PubMed.
- S. Deechongkit and J. W. Kelly, J. Am. Chem. Soc., 2002, 124, 4980–4986 CrossRef CAS PubMed.
- F. Delom and E. Chevet, Proteome Sci., 2006, 4, 1–12 CrossRef PubMed.
- I. L. Batalha, C. R. Lowe and A. C. A. Roque, Trends Biotechnol., 2012, 30, 100–110 CrossRef CAS PubMed.
- L. Ashton, C. Johannessen and R. Goodacre, Anal. Chem., 2011, 83, 7978–7983 CrossRef CAS PubMed.
Footnotes |
| † Electronic supplementary information (ESI) available. See DOI: 10.1039/c4ra16934d |
| ‡ Previous address: Instituto de Tecnologia Química e Biológica António Xavier, Universidade Nova de Lisboa, 2780-157 Oeiras, Portugal. |
| This journal is © The Royal Society of Chemistry 2015 |