
Facile synthesis of copper nanoparticles in glycerol at room temperature: formation mechanism†
Abstract
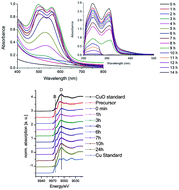
A copper sol is usually synthesized by the reduction of a copper precursor with a suitable reducing agent in the presence of a stabilizer. There are few reports regarding the preparation of copper nanoparticles in glycerol without using a stabilizing agent, but at elevated temperatures. The formation of reduced copper (Cu0) is usually verified by a UV-vis spectrophotometer where a ‘red copper sol’ was formed. In the present paper we synthesized the copper sol at room temperature in a glycerol medium using hydrazine as a reducing agent. The chemical states of copper in the sol and their composition were analyzed by X-ray absorption near edge structure spectroscopy (XANES) with the linear composition fitting method. A series-parallel mechanism of the reaction was proposed. An average particle size of 5 ± 1 nm was visualized via transmission electron microscopy (TEM).
 Please wait while we load your content...
Please wait while we load your content...

