
Photomagnetic molecular and extended network Langmuir–Blodgett films based on cyanide bridged molybdenum–copper complexes†
Abstract
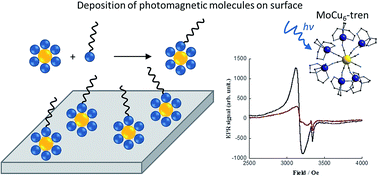
Two types of cyanide bridged molybdenum–copper photomagnetic films have been obtained: the first one is based on a molecular [MoCu6] complex, the other being a two-dimensional [MoCu2] coordination network. Both systems employ surfactant functionalized ligands and films were deposited on Melinex substrates using the Langmuir–Blodgett technique. All systems, including monolayer films, showed full retention of the intrinsic photomagnetic properties known for analogous solids as demonstrated by EPR spectroscopy.
- This article is part of the themed collection: Luminescence and photophysical properties of metal complexes
 Please wait while we load your content...
Please wait while we load your content...

