
Double-edged sword in cells: chemical biology studies of the vital role of cytochrome c in the intrinsic pre-apoptotic mitochondria leakage pathway
Zhi-Peng Wangabc,
Xiao-Zhe Dingb,
Jun Wang*a and
Yi-Ming Li*a
aSchool of Medical Engineering, Hefei University of Technology, Hefei, Anhui 230009, China. E-mail: wangjun-08@126.com; lym2007@mail.ustc.edu.cn; ymli@hfut.edu.cn
bDepartment of Chemistry, School of Life Sciences, Tsinghua University, Beijing 100084, China
cChemistry Department, Texas A&M University, College Station, Texas 77840, USA
First published on 11th March 2015
Abstract
Besides functioning as an electron transporter in the mitochondrial electron transport chain, cytochrome c (cyt c) is also one of the determinants in the execution of cell death. The interaction between cyt c and lipids has long been of great interest in biological systems, while the study of cyt c has attracted even more attention since its new function in apoptosis was discovered. Theories on the cause-effect signaling pathways have been proposed. However, until recently, some of the detailed parts have remained poorly understood in the big picture of cyt c-mediated apoptosis. In the past few years, new labelling, monitoring and detecting methods as well as in vitro model systems such as cyt c–cardiolipin (CL) or cyt c–membrane systems have been developed to overcome these drawbacks. The discovery of the versatile roles of cyt c in metabolic as well as apoptotic processes suggests cyt c per se as a potential drug target. In this review, we divide the whole cyt c-mediated pre-apoptotic mitochondrial leakage process into several sections in a chronological order and summarize the recent discoveries and hypotheses. With the combinatorial effort of modern interdisciplinary subjects such as chemical biology, bio-inorganic chemistry, structural biology and physical organic chemistry, we expect that researches in this field will shed light on our understanding of the whole intrinsic apoptotic process, and further contribute to health sciences.
1. Introduction
1.1 Background for cyt c
Cyt c is a mini-hemoprotein which was first characterized in the early 1920s.1 The first identified role of cyt c was as an electron transporter in the oxidative phosphorylation pathway.2 Cyt c shuttles electrons between complex III (cyt bc1) and complex IV (cyt c oxidase) in mitochondrial intermembrane space (MIS), which can be described by the random collision model.3 To achieve normal functions as a transporter, cyt c is maintained at a high concentration of 0.5–1.0 mM in MIS (Fig. 1).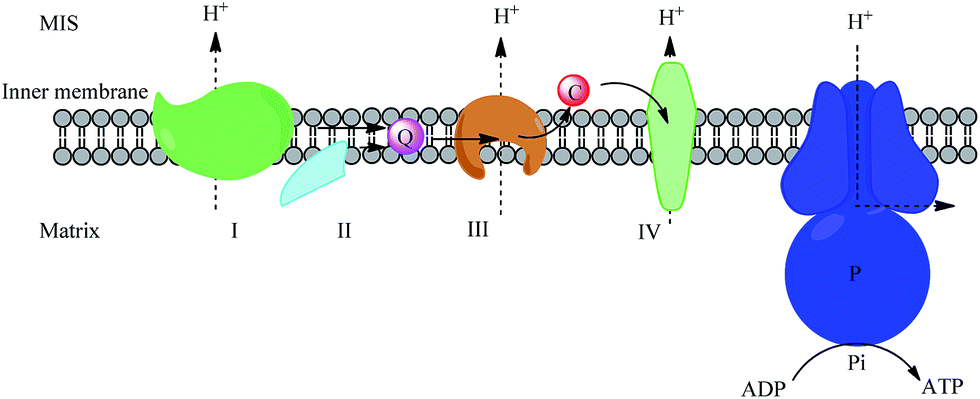 | ||
| Fig. 1 Schematic of the mitochondrial electron transport system in the inner mitochondrial membrane (IMM). I, II, III and VI stand for complex I (NADH dehydrogenase or NADH:ubiquinone oxidoreductase), complex II (succinate dehydrogenase), complex III (cyt bc1) and complex IV (cyt c oxidase) respectively; Q stands for ubiquinone; C stands for cyt c; P stands for ATP synthase. | ||
Cyt c shares common properties with other hemoproteins: a heme prosthetic group (together with its redox intermediate) forms a covalent bond with the protein backbone via thioether bonds. An iron ion as the center metal coordinates with heme as well as His 18 and Met 80 residues, to reach its six-coordination structure. Iron oxidation/reduction between the ferrous and ferric states is known to endow the protein with an electron-transferring function. The c-type linkage of heme combined with the axial ligation is important to increase the heme reduction potential. Besides the Fe–heme structure, the overall structure of cyt c is a globular folding structure with five α-helices and loops well-packed around the heme group.4 There are three long α-helices and two short α-helices (Fig. 2). The folding mechanism and folding kinetics of cyt c in vitro have been well-studied,5 and the folding of cyt c was utilized as a typical model system for the research of how unfolded proteins renature.6
 | ||
| Fig. 2 Structure of horse heart cyt c 1-104 with heme (Protein Data Bank (PDB) code 1HRC): the left panel shows the crystal structure of cyt c; the right panel is the protein topology diagram (Pro-origami)7 showing the five α-helix secondary structure. | ||
1.2 Role in apoptotic signaling pathway
Apoptosis is programmed cell death, which plays an important role in all stages of the development of an individual organism, such as the development of the embryo, elimination of unnecessary cells as well as the maintenance of the physiological balance. Cys-dependent Asp-specific proteases (caspases) are a group of cysteine proteases which are the main executioner of substrate cleavage. Two key pathways in vertebrates can activate caspases: (1) the intrinsic and (2) the exogenous apoptosis pathways.8 DNA damage, metabolic stress or the presence of excess amounts of unfold proteins can all be stimuli for the intrinsic apoptotic signalling pathway or the mitochondrial pathway. These stimuli can pass a signal to the mitochondria system and cause the pre-apoptotic signalling cascade, and finally lead to the formation of the apoptosome. The apoptosome can subsequently activate caspase-9 to cleave the key apoptotic factors caspase-3 and 7, and lead the cell to an apoptotic fate.9Although the structure and properties of cyt c have been studied in detail for more than 70 years, its important role in the pre-apoptotic signalling pathway, as its second major role in cells, was not discovered until recently. In 1996, Wang’s lab10 first proposed that cyt c released from the mitochondria might be of vital importance to the intrinsic apoptotic pathway in a cell-free extract system. Four years later, Williams and collaborators11 confirmed this conclusion by several important in vivo experiments.
Now, a general picture of the role of cyt c in apoptosis has been generated: cyt c normally binds to an anionic phospholipid CL specifically existing in the inner mitochondrial membrane, and the release of cyt c from mitochondria executes the downstream apoptotic process. Mitochondrial synthesis of reactive oxygen species (ROS) is one of the key factors that triggers the release, which contributes to an increase of peroxidase activity and CL oxidation. Since ROS are commonly regarded as harmful factors to cells, the increasing level of ROS has also been considered as a significant signal for the induction of apoptosis. Thus, oxidation of CL might be necessary to induce mitochondria membrane leakage, and the release of pro-apoptotic substances, such as protein tyrosine phosphatases (PTP), into the cytosol. Additionally, the released cyt c can play some other roles in apoptosis. It binds to the apoptotic protease-activation factor-1 (APAF1), and stimulates its heptamerization. Afterwards, cyt c, APAF-1, and ATP/dATP will assemble into the apoptosome for the further activation of pro-caspase-9. With the help of the apoptosome complex, caspase 9 gains the ability to trigger the further caspase signal downstream to finally activate caspase-based apoptosis. Additionally, the apoptosome will also stimulate the extracellular matrix-digesting metalloproteinase 9 (MMP-9), and the gene expression of inflammatory cytokines will also significantly increase.12 It also interacts with and then oxidizes another negatively charged phospholipid phosphatidylserine (PS).13 Many studies have reported on the cyt c-lipid interaction and will be discussed later in the review. However, some detailed questions remain unknown and several current discoveries or hypotheses are about to shed light on these mysteries.
1.3 Cyt c, CL and mitochondria
There are three key players in the early initiation stage of apoptosis: cyt c, CL and mitochondria, which are closely related to each other. Mitochondria provide a necessary and indispensable place for the interaction between cyt c and CL. On the one hand, cyt c gets its fully folded structure in mitochondria, and the entire life cycle of cyt c is tightly connected with mitochondria. According to endosymbiotic theory, mitochondria, together with hydrogenosomes and plastids, came from free-living prokaryotic cells to endosymbionts of eukaryotic cells via endocytosis.14 Therefore, the DNA and ribosome systems remaining inside are able to express some peptide chains independently. However, some mitochondrial proteins, for example the apoprotein of cyt c from different species,15 are actually encoded by chromosomal DNA.16 Regarding its biosynthesis in vivo, after being translated to a polypeptide chain at the endoplasmic reticulum or microsome,17 cyt c is transported into the mitochondria to assemble and obtain the necessary heme molecule. Neupert and Hennig18 provided evidence to demonstrate that cyt c pass through the outer mitochondria membrane (OMM) with the help of certain kinds of receptors in a cell-free system. In MIS, an enzyme cytochrome c heme lyase (CCHL) catalyzes the formation of thioether bonds between heme and the apoprotein (Fig. 3). Besides heme reconstitution, no other post-translational modifications (PTM) on cyt c have been reported.19 It should also be mentioned that the mitochondrial metabolism sophisticatedly regulates the expression level of cyt c, but has no significant influence on its stability.20 | ||
| Fig. 3 Schematic for cyt c bio-synthesis in the cytosol, and assembly in the mitochondria. | ||
Interestingly, only holo-cyt c released from mitochondria (but not the apoprotein that has not been transported into the mitochondria) can induce apoptosis. In other words, cyt c in its apoprotein form is not an apoptotic signalling molecule, while the holoprotein form is. A potential reason for this phenomenon is the role of the iron center. The mechanism of this phenomenon is as follows. Metalloproteins often do not fold unless they are in the presence of cofactors or prosthetic groups, and this principle is applicable to cytochrome proteins. Without binding to heme groups, apocytochromes tend to have no stable structures. These states were named as molten globule states.21 This may be able to explain the above-mentioned phenomenon that the holo-form of cyt c can be a signalling transferer only with the incorporation of a heme group, in other words in a form with oxidase activity. This mechanism also has some potential therapeutic applications. If we can manually incorporate heme or its derivates into apo-cyt c and let it fold in the cytosol, we might be able to induce “mitochondria-free” intrinsic apoptosis. This would provide a potential strategy to kill undesirable cells inside the body.22
On the other hand, mitochondria provide an irreplaceable and rather closed surrounding for cyt c–CL interaction to take place in the whole pre-apoptotic pathway. CL is a family of phospholipids with two negative charges (Fig. 4). Since there are four fatty acid moieties inside each CL molecule, this family contains large numbers of variants. However, both the amount and the types of CL are varied in different species, cells and membranes, which is still unexplainable so far.23 For example, early studies24 have shown that around 25% of all the mitochondria lipids under physiological conditions are CLs. Furthermore, there is an unbalanced distribution between the IMM and OMM in which more than 65% of CLs are inset into the IMM.25 It is worth noting that all the enzymes used in primary CL biosynthesis are inside the mitochondria: the whole pathway starts from an enzyme in the OMM26 to enzymes in the IMM27 and ends on the mitochondria matrix side of the IMM.28 This physiological situation of CL distribution will be changed when several CL-binding proteins29 interact with CL as well as its metabolites, which will cause CL enrichment in the OMM. The distributional change will facilitate cyt c release from the mitochondria into the cytosol and further induce apoptosis.
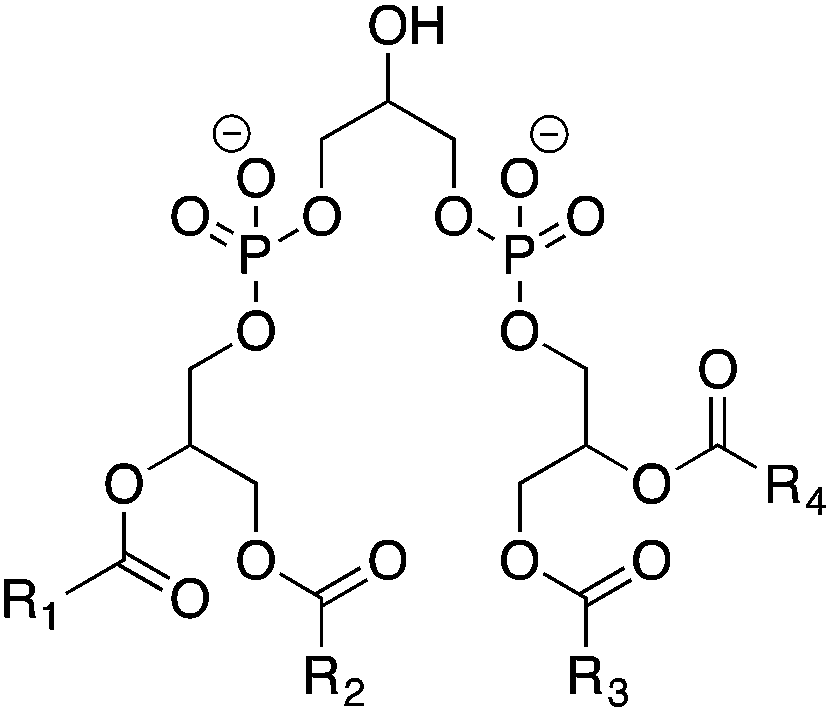 | ||
| Fig. 4 Chemical structure of CL family lipids. | ||
2. Binding to lipids & oxidation of lipids
Besides the key electron transferring role in the oxidative signaling pathway, cyt c has recently been found to play a signal mediator role in the intrinsic pre-apoptotic pathway.30 The interaction with CL is believed to be a vital factor for this step.2.1 Binding to lipids and lipid containing membranes
It has been a very long time since scientists started studying cyt c–lipid interaction. Peptide analysis demonstrated that its essential structural characteristics endow the reactivity. Cyt c has an isoelectric point (pI) near 10, with a +8 charge at neutral pH. Thus, it has a high tendency to bind to negatively charged molecules such as CLs. Cyt c in its native structure can interact with certain kinds of lipids, such as catalyzing the fatty acid amidation reaction.31 The fact that cytochrome c can interact with the lipid bilayer has been known for tens of years,32 even before the term “apoptosis” was defined.As a result, scientists became interested in the binding mechanism between cyt c and lipids or the membrane ever since its discovery. Many studies have been published based on different model systems,33 and these studies have demonstrated that cyt c can bind to the lipid layer strongly through electrostatic interactions with the negative charges.34 Besides ionic interactions, researchers35 also showed that there are hydrophobic interactions between cyt c and the lipid bilayer. For example, the ionic strength increase or the dilution of the cyt c concentration cannot dissociate the protein from the membrane. Additionally, the ionic strength is exactly suitable for cyt c to show persistent binding under physiological conditions.36 In contrast, the initial binding of cyt c to either CL or the phosphatidylglycerol (PG)-containing membrane is sensitive to both of these two factors.37 Some other observations even made the process more complex: Marsh and Heimburg38 applied physical chemistry in the protein–protein interaction study, and the binding isothermal analysis showed that there might be more than one way for cyt c to interact with the membrane: a competition between an “out-of-plane” membrane binding mode with an “in-plane” membrane binding mode to differentiate the relative relation of cyt c to the membrane. Pinheiro39 tried to explain this by providing two hypotheses in his review article. Firstly, cyt c can have an initial interaction with the membrane mainly through an electrostatic interaction, and the electrostatic interaction nature causes small changes in the lipid bilayer. At this stage, cyt c only slightly penetrates into the membrane. However, 31P NMR shows that the binding with cyt c will cause the generation of non-bilayer structures or lateral phase separation of enriched acidic phospholipids, especially in cardiolipin bilayer regions. At the same time, ESR or resonance Raman spectroscopy suggests that cyt c, upon binding with lipids, will undergo a backbone structural change around or within the heme part. Followed by the changes of both the protein structure and the lipid components, the dynamic characteristics of the membrane-binding cyt c will significantly change: no helix structure can be detected with a life time over 10−6 s, which suggests a different binding model.39 Two years later, with the use of steady-state surface plasmon resonance (SPR) spectroscopy for the binding study over different conditions, Tollin40 demonstrated that cyt c can bind to the lipid membrane by two phases with different binding constants. These results verified the hypothesis that there is an initial binding through pure electrostatic interactions followed by a secondary binding through hydrophobic interactions. In contrast, Pinheiro and coworkers41 even discovered and characterized five possible protein conformations in the cyt c–membrane interaction system. Much effort has been put into the detailed study of CL–cyt c binding,42 however, this mysterious phenomenon is waiting to be further deciphered.
In the current theory, the result of the binding is twofold. On the one hand, Cullis and De Kruijff33 found that the binding of cyt c can reversely change the CL-containing membrane structure to form an “inverted hexagonal phase” as well as an inverted micellar structure, which was an important discovery. Demel et al.43 took this one step further on a systematic scale. They let cyt c in both the apo-form and holo-form interact with membrane extracts from different organelles and measured the differences of the binding results. They tried to rationalize from a biological point of view that acidic lipids could play a regulatory role in triggering the importation of cyt c into the mitochondria. On the other hand, the binding can also affect the protein vice versa besides causing increased oxidation activity, which results in the membrane changes. We will discuss induced oxidation activity here, and the biophysical and kinetic studies will be discussed in the third section. A conformational change of the protein and a structural change of the membrane have also been reported. Hoch’s group44 reported the binding of cyt c to acidic phospholipids such as the negative charged CLs. This binding can cause protein structural changes in its immediate surroundings as well as the heme group’s coordination model (discussed in the next section). In addition, Surewicz and coworkers45 found that the protein’s main chain structure is even considerably altered from a global folding structure to a more extensive form, which means that binding to the membrane can make the protein structure unstable. After this, a cohort of effect was devoted to the protein’s structural characterization. How can this interaction have such a huge impact on the protein structure? Englander’s group46 found that among all the folding domains (so called “foldon”), the 40s Ω loop with the amino acid sequence from 40 to 57 (ref. 47) is the least stable. It is potentially stabilized via forming hydrogen bonds of His 26 to Pro 44 and Thr 49 to the heme propionate.48 In contrast, if the hydrogen bonds are interrupted, this foldon folding will be significantly weakened, such as under acidic conditions when protonation can break the hydrogen bonds.49 The disruption of the Ω loop can even have greater effect on cyt c conformation. Spiro and Groves50 used Resonance Raman spectroscopy (RR) to discover a totally novel structural change to β sheets when heating cyt c under acidic conditions. Only part of the cyt c could undergo this transformation, while the rest of the proteins just unfolded. They explained this finding with the hypothesis that the unfolded 40s Ω loop (accounting for more than 1/3 of the amino acids) can extend to an additional β strain structure, which results in the huge transition to an entire β sheet structure. Jemmerson’s group51 reported in 1999 that cyt c could have a Pro 44-related conformational change after binding to the artificial phospholipid-containing vesicles. In that research, they used a monoclonal antibody specific for the unfolded domain around Pro 44 as the detection tool, and found that it only bound to cyt c when it was interacting with an artificial phospholipid membrane. Furthermore, this result was confirmed by the in vivo data of T hybridoma cells in the post-apoptotic stage.
Another important finding was the structural change in the heme group during the interaction. Faljoni-Alario’s group52 reported that the interaction between cyt c and lipids could significantly change the heme group binding model as well as the iron coordination state. Hildebrandt and coworkers53 studied the heme spectroscopic changes upon this interaction, and provided a binding theory to rationalize it. The change of the heme group together with its subsequent effects will be discussed in the next section.
To summarize, the binding of cyt c to CL causes significant changes to the protein structure, and plays an important role in the apoptotic pathway.13 This interaction model has also been regarded as a paradigm for peripheral membrane proteins38 by theoretical chemists for the following reasons. Firstly, cyt c can be an easily-isolated enzyme and has been well characterized, and secondly, its binding with negatively charged phospholipids in the membrane system is very strong, which could be relatively easy to study.33
2.2 Oxidation of lipids
Knowing that cyt c undergoes dramatic conformational changes at both the secondary and tertiary levels upon binding with CL, people became curious about the functional effects corresponding to it. In general, the structural rearrangement of the protein can interrupt the sixth coordination of Met 80 to the Fe–heme, and thus lead to an exposed heme structure. At the same time, this penta-coordinated iron can also be open to other ligands or substances. However, under certain situations, the high-spin iron with penta-coordination might drag His 33 as an alternative to form a hexa-coordinated structure. It should also be mentioned that in the native form, the intrinsic Trp 59 fluorescence is quenched by the Fe–heme center. This protein conformational change can remove the quenching effect of heme. Therefore, Trp 59 fluorescence appears to provide an important labelling tool for further cyt c study.54Large amounts of evidence show that it is CL peroxide instead of CL per se that plays a role in the membrane leakage.55 However, the generation mechanism of CL peroxide is unclear. In 2005, Kagan and coworkers56 made use of the oxidative lipidomics technique to find out that cyt c could specifically oxidize CL to the peroxide after binding to CL on the membrane. This result is important for two reasons: (1) we finally know that the increase of cyt c peroxidase activity is also tightly related to its own leakage to the cytosol. It is also very surprising that only CL can be oxidized in the presence of many other saturated and unsaturated phospholipids in vivo. (2) This work shows the necessity for reactive oxygen species in the apoptosis pathway, which provides us with an understanding of the triggering role that they play. In 2006, our knowledge of the importance of the peroxidase activity advanced one step further. Kagan’s group57 verified that the hydroperoxide product from the cyt c oxidation of CL was actually necessary for the releasing of the proteins. H2O2 is known to be necessary. The peroxidase activity can even be activated under lower H2O2 concentrations when binding to CL than the activation of its native form.
Later, it was found that CL is not the only kind of lipid that can be oxidized at certain conditions in certain cell lines.58,59 Actually, a small group of lipids, including PS and phosphatidylinositol, can also be oxidized, and the products have systematically studied using mass spectra (MS).58 There are two important kinds of negatively charged lipids among all the lipids oxidized by cyt c. One is CL at the early apoptotic stage inside the mitochondria, and the other is PS after the release of cyt c.59 Different CL oxidation products have been identified in apoptosis-induced mice cells. Most of them are CL with different levels of peroxidation on the fatty acid chains (short for CL–OOH) including (C18:2)3/(C18:2–OOH)1, (C18:2)2/(C18:2–OOH)2, (C18:2)1/(C18:2–OOH)3, and (C18:2–OOH)4. The cyt c oxidation product, mostly peroxide or oxide, might be related to a CL hydrolytic reaction. This suggests a cyt c-catalyzed CL remodeling, and a potentially new physiological function of cyt c.54
Integrating all the information at hand, we would like to summarize the big picture of the cyt c–CL binding-induced pre-apoptotic signaling pathway (Fig. 5). Under normal conditions, CL is mostly localized in the IMM, and cyt c plays the primary role of electron carrier between complex III and IV in the MIS. However, under non-physiological conditions, CL is induced by some apoptotic signals to rearrange to a biased distribution between the IMM and the OMM. This rearrangement will cause cyt c to go through a series of changes to get to another edge of the sword, including: (1) a relocation of cyt c to the OMM, (2) a conformational change of cyt c, (3) an increase in the peroxidase activity of cyt c, (4) peroxidation of CL by cyt c to break the membrane and finally (5) the leaking of cyt c to the cytosol. The whole story of the cyt c–CL interaction is quite clear now; for more detailed information about the cyt c–CL interaction as well as the role of cyt c in causing intrinsic apoptosis see other reviews.54,60 Remarkably, lots of different stimuli can cause apoptosis, such as the channel forming protein Bax,30 and thus there might be different signaling pathways corresponding to each of them. Orrenius and coworkers61 proposed the Bax-assisted cyt c leakage theory. Bax is known as the most effective trigger reagent to make pores in the membrane and cause around 19% cyt c release from the mitochondria. In this study, they found that in Bax-treated mitochondria systems, the release of cyt c to the cytosol contains two steps: (1) the re-solubilization of cyt c from the CL-bound form, and (2) a Bax-assisted membrane-permeabilization. In that case, the strong interaction between cyt c and CL does not assist, but reversely hinders, the cyt c leakage. To date, we still lack detailed information on the conformational change as well as the membrane leakage process. Some important discoveries further shed light on the mystery, which will be discussed in later sections.
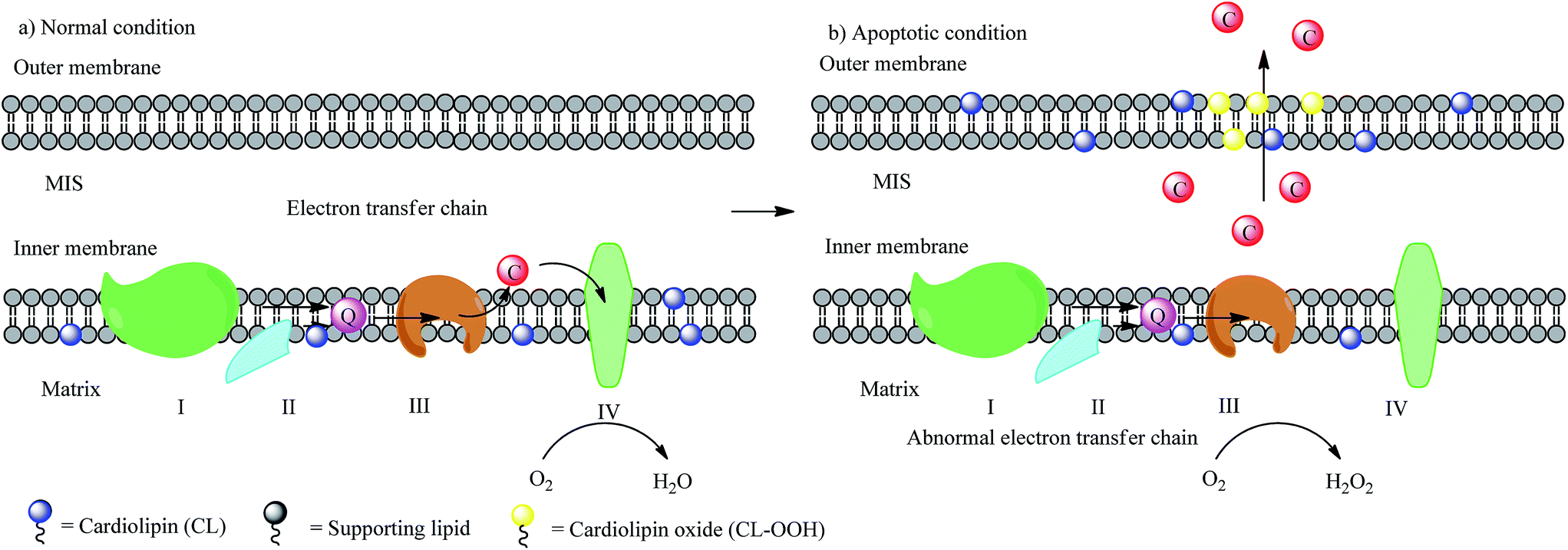 | ||
| Fig. 5 Scheme of CL oxidation-induced membrane leakage. I, II, III, and VI stand for complex I, II, III and IV respectively; Q stands for ubiquinone; C stands for cyt c. | ||
3. Conformational change
Binding of cytochrome c with CL causes partial unfolding of the protein. Following the binding to CL, cyt c undergoes an important conformational change between the compact (C) and the extended (E) protein conformers. The rate for this exchange process is highly related to the degree of unfolding as well as cyt c interacting with CL.3.1 Conformational change to an extended form
Although a large body of researches have showed that cyt c can undergo a conformational change which leads to an increase in peroxidase activity,57 the unfolding process and detailed structure of the CL-bound cyt c is still unknown to the field. The reason why we are not able to overcome this challenge was largely due to the lack of efficient labelling and analyzing strategies. As mentioned above, in the early stages Trp fluorescence was used as a native probe for protein folding kinetics studies.62 What kind of conformational changes occurred to cyt c is a question that long puzzled researchers, until the introduction and application of the dansyl (Dns)-labelling method into this field to provide a glimpse into the molecular level. The Dns-labelling method has been proved to be an important and useful weapon for the hemoprotein biophysical study of protein folding, protein–ligand interaction, enzymatic mechanism, etc. In this strategy, dansyl groups are utilized to label a series of target protein variants with different mutant Cys sites. Thus, fluorescence energy-transfer kinetics (FRET) is applicable to analyse an important factor, dansyl-to-heme distance distribution [P(r)], to give structural information (Fig. 6).63 This important structural tool has been applied for the cyt c folding studies.64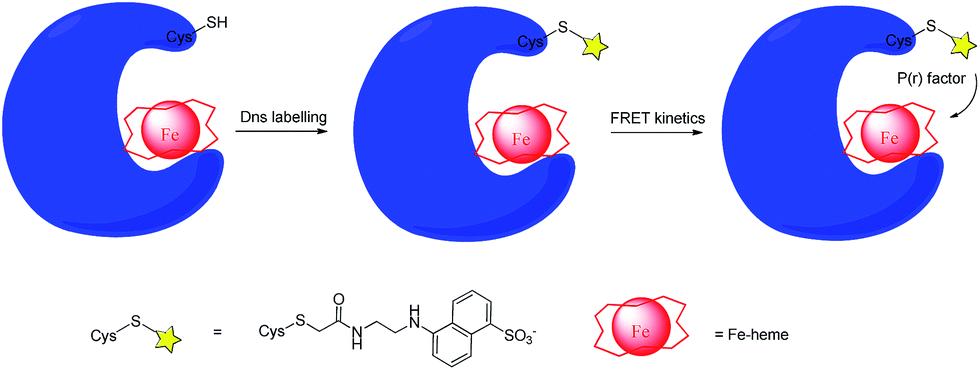 | ||
| Fig. 6 Scheme of the principle of the Dns-labelling method and further FRET kinetics study. The Dns group is shown as a star. | ||
Combining this labelling technique and time-resolved FRET, Pletneva and co-workers did a series of structural researches on cyt c bound to CL, as discussed below. All these processes are not easy to study with classical methods and probes. They created four horse heart cyt c variants at electrostatic binding positions (E4C, K39C, E66C, and E92C) to study their Dns decay curve–P(r) relation.65 The results showed that different variants undergo diverse protein conformational changes to degrees and populations based upon the CL–cyt c interaction. Among all these CL binding cyt c ensembles, one of the subpopulations with a largely opened conformation seemed to play a dominant role in the peroxidase activity (Fig. 7). Additionally, the results also demonstrated the key role of electrostatic interactions between CL and cyt c, especially at the protein C-terminal α-helix domain.
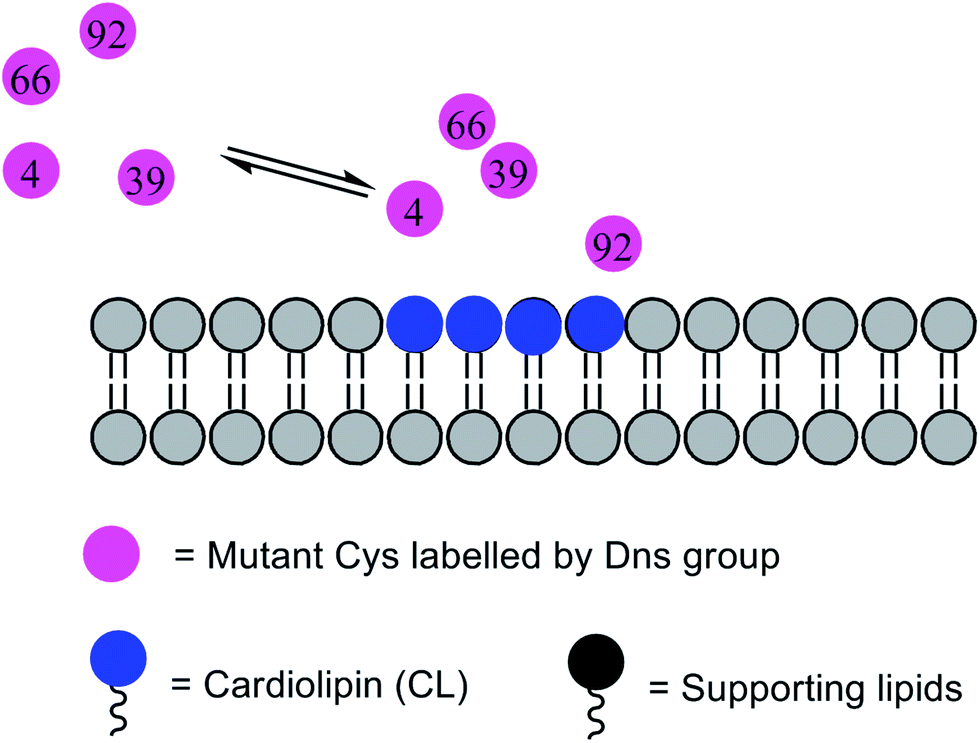 | ||
| Fig. 7 Scheme of the conformational change of cyt c after binding to CL. | ||
In 2012, Pletneva’s group66 went one step further on the structural characterization. Besides time-resolved FRET and the Dns-labelling method, they introduced two other techniques, biolayer interferometry (BLI) and fluorescence correlation spectroscopy (FCS), into this realm, and demonstrated their important roles in kinetic investigations. With the help of these techniques, Pletneva et al. induced another labelling group, Bim, to the Glu 92 site in order to study the protein biophysics more systematically. They studied the equilibrium between the compact form and the extended form, the exchanging rate between two conformations, as well as the protein–CL electrostatic interaction kinetics. They found that the rates of cyt c binding to the membrane and the subsequent conformational change were largely related to the concentration change of CL. Based upon these understandings, they proposed a peripheral interaction mechanism that made a major contribution to the denaturation of cyt c after binding to the CL surface. These works also showed that the development of novel powerful techniques could largely facilitate protein biophysics studies. Some of the other important new developments such as single molecule studies with shorter time scales67 are all potential weapons for taking protein–ligand interaction research one step further.
Homologous proteins from other organisms have also been studied. Pletneva et al.68 studied the structure and oxidation activity of two recombinant expression cyt c proteins, CYC-2.1 and CYC-2.2, from one nematode species, Caenorhabditis elegans. They found that both of the two proteins could interact with CL-containing liposomes, and that detectable peroxidase activity was similarly induced by the interactions. Afterwards, they applied their biophysical tools to this system to discover the partition of the compact form and the extended form, which is again similar to mammalian systems. Combined with a huge number of cellular biology and genetic studies of the C. elegans system,69 the result suggested that nematode cyt c proteins might be a potential helpful model system for further in vitro and even in vivo studies.
Besides the conformational changes discussed above, some other changes have also been reported. It should be mentioned that Hirota and coworkers70 reported that cyt c could oligomerize or polymerize through successive domain swapping under in vitro conditions. In the oligomeric structures, the C-terminal α-helix of one protein is displaced by the C-terminal α-helix of another protein which leads to a significant perturbation of the Met–heme interaction. The weakened coordination between Met and heme could cause higher peroxidase activity in dimeric cyt c compared with the monomer form. This discovery suggests that the C-terminal α-helix is a relatively separated folding moiety to the cyt c body.
3.2 The effect of ATP
Adenosine 5′-triphosphate (ATP) is one of the vital components in mitochondria,71 and close attention has been paid to ATP under physiological conditions. For example, the concentration of ATP is an important indicator for the mitochondria and cell activity, and different methods have been applied to measure the cellular ATP level.72 The interaction of ATP with cyt c is complex but also very intriguing.73 Early in vitro evidence showed that a low concentration of ATP is necessary for the activation of the apoptosome,74 and in vivo studies have shown that extracellular ATP can even trigger apoptosis.75 The mechanism for apoptosis induced by extracellular ATP76 has also been revealed, which turned out to be ATP-caused reactive oxygen species (ROS) generation. Since 2001, ATP has been found to induce structural changes on cyt c, which is important for the apoptosis pathway.77 However, higher levels of ATP can inhibit the apoptosis.78 The binding of cyt c to negatively charged lipids or fatty acids can cause the unfolding of the protein. However, Fiorucci and coworkers79 found that only ATP among all the nucleotides can help to turn cyt c back to the folded form. This was a new function of ATP in mitochondria, aside from being a product from the oxidative chain.To summarize, ATP cannot only bind to cyt c per se, but also regulate the possible conformational change of cyt c caused by the significant interaction between CL and cyt c. The interaction between cyt c and CL is mainly ionic,77 and thus it is highly related to the ionic strength of the surrounding environment. The interaction can even be broken by high ATP levels, similar to the effect under high sodium chloride concentration.80 Additionally, some other phenomena in similar systems also provide relevant information. Oleic acid is known to bind to cyt c,79 which will result in the destabilization of the protein. However, ATP is known to feature the capacity to weaken this unfolding effect to some extent, not only for oleic acid-bound cyt c, but also fragment complex 1-56/57-104 residue of cyt c (also known as nicked cyt c), and even acid -denatured cyt c.81
There are also some theoretical models of the ATP–cyt c interaction. Pletneva and co-workers66 have demonstrated that the balance between a compact protein form (C) and an extended form (E) is determined by the degree of protein unfolding as well as the cyt c–CL interaction. They82 used five different dansyl (Dns)-labelled cyt c variants as models to show that both ATP and salt can inhibit the interaction between cyt c and CL-containing liposomes. However, besides simply competing with the phosphate groups of the lipid, ATP can additionally increase the peroxidase activity of the CL-bound cyt c. Based on the structural information, they further proposed an innovative explanation for the conformational change pathways, in which ATP plays an important regulating role. ATP raises the population of E through destabilizing a native-like compact species C′ (Fig. 8). It should be mentioned that GTP can play a similar role in affecting the conformational change, showing that cyt c has a high tolerance over different nucleoside moieties.
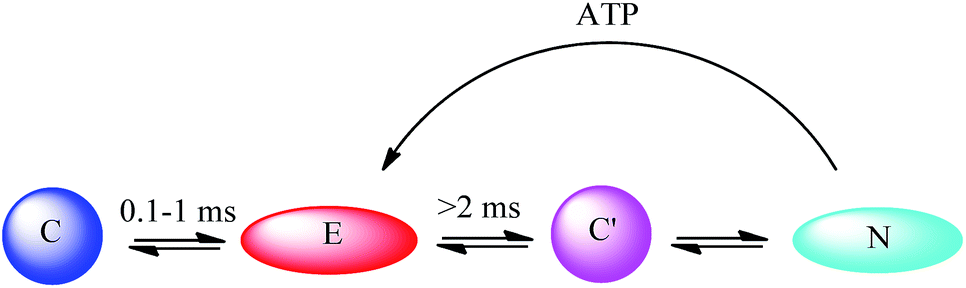 | ||
| Fig. 8 Conformational change pathway proposed by Pletneva and co-workers.66 C stands for the compact form; E stands for the extended form; C′ stands for another compact form; N stands for the unfolded form. | ||
Another important finding related to the phosphate group is that cyt c can undergo phosphorylation conveniently. Since this process can add one negative charge to the system in total, it might block the interaction of cyt c with anionic membrane lipids. Several researches have reported the existence of a Tyr 48 (ref. 83) variant in liver cells and a Tyr 97 (ref. 84) variant in heart cells. Whether this is related to ATP is also unclear.
To summarize, the current understanding provides some general information about the interaction between cyt c and CL as well as ATP. Those structural details, such as binding sites, interaction models, and even the stoichiometry between the protein and the small molecules, need to be further discovered and verified. Different biophysical tools are all applicable to this open field, for example, deuterium NMR is a traditional tool which has long been used to study cyt c and its interaction with the membranes;85 Fourier transform infrared (FTIR) combined with amide H-exchange has also been used to study the secondary and tertiary structural changes of cyt c.86 Recently, Roder’s group87 used ultrafast NMR-detected H/D exchange experiments to study the folding kinetics of cyt c. They found that both N- and C-terminal α-helices could fold in a later time period compared with the other parts of the protein. This also suggests a rather weak interaction between the N-terminal α-helix and its body, as well as the independence of the N-terminal α-helix’s folding.
4. Membrane leakage
It has been a long time since cyt c-CL-containing membranes were closely investigated with model membrane systems, and different facilities have been used for the read-out.88 However, these studies did not provide information about a more detailed process of membrane leakage before the content release. A new model system was recently developed to give us more solid information.4.1 Giant unilamellar lipid vesicles (GUVs) model system
It is already known that the binding between cyt c and CL in the mitochondrial membrane can result in a protein conformational change to an extended form, as well as the increase of peroxidase activity. This is one of the key steps in the intrinsic apoptotic pathway. What influences this protein–lipid interaction on the membrane remains an unsolved problem. Giant unilamellar lipid vesicles (GUVs) have been applied as model systems to many biological researches such as protein–membrane interactions and membrane permeability.89 New methods are being developed to help us to rapidly synthesize GUV from various phospholipids.90 Groves and collaborators91 made use of this simplified synthetic bio-membrane system with separated phases of CL-rich domains. One of the advantages of using GUVs is the visibility of both inside and outside solution conditions via laser scanning confocal fluorescence microscopy. With the help of the novel system, they observed that the cyt c–CL interaction could induce twofold membrane structural changes. On the one hand, the CL-rich domain of a single vesicle could give out buds and end up in a morphologically collapsed form. On the other hand, the CL-rich areas between several adjacent vesicles could make the membrane deform to form a complex osculating region (Fig. 9). Additionally, they found that other proteins or macromolecules with multiple positive charges and similar sizes were all able to show qualitatively similar phenomena. This suggests that there is a colloidal force inside this bi-component system. This mitochondrial related protein–membrane interaction study could provide a better understanding of the establishment of cristae structures of the IMM under physiological conditions, as well as the membrane leakage during apoptotic conditions. | ||
| Fig. 9 Effect of cyt c and CL interaction to the membrane. (a) Effects on single GUV; (b) effects on multiple adjacent GUVs. | ||
Another important question that remains unanswered is how cyt c, together with other mitochondrial proteins, is released into the cytosol. To find out whether cyt c penetrates the membrane independently, Groves’ lab92 established a model system to study fluorescent cyt c interacting with artificial GUVs containing CL without the existence of any other proteins. Using this directly visible system, they observed the binding of cyt c to the membrane, and the leakage of cyt c out of the membrane. They demonstrated that the formation and maintenance of membrane pores is achieved solely by cyt c. Not only cyt c itself, but also some fluorophores, such as carboxyfluorescein, as well as the macromolecule dextran with a 10 kDa molecular weight (MW), can pass through the membrane. However, the size of the pores is limited, since larger dextrans cannot cross. Another interesting finding was that ATP would largely decrease the formation of pores and sequential leakage. Thus, a CL-induced membrane pore formation and mitochondrial content leakage pathway was proposed (Fig. 10).
 | ||
| Fig. 10 Cyt c and CL interaction cause membrane leakage. | ||
4.2 The potential role of C-terminal α-helix
Based on the materials at hand, Groves’ lab92 highlighted the role of the C-terminal α-helix in the formation and stabilization of opening the membrane pores. Although this has not been fully verified, several hints are all pointing to this hypothesis.Firstly, with bioinformatic methods, they found that the C-terminal α-helix contains a highly conserved sequence, KKEERAD, starting from Lys 88, and helical wheel analysis (Homo sapiens: KKEERADLIAYLKKATNE) reveals an aggregation of positive charges on one helical face, with negative charges on the opposite end (Fig. 11). The multiple net positive charges can highly facilitate the protein’s interaction with CL, and the negative charges might be able to drag other proteins as well as other materials with positive charges through the pores.
 | ||
| Fig. 11 Helical wheel analysis of the C-terminal α-helix, axle direction from N to C: into page. | ||
Secondly, combined with the structural studies by Pletneva and coworkers, the unfolded C-terminal α-helix, which extends from the body of the protein after the conformational change, has a high possibility of inducing pore formation and maintenance. Additionally, Arg 91 (ref. 73) is one of the key conserved amino acid residues in the C-terminal α-helix which plays an important role in cyt c peroxidase activity. The Worrall group93 demonstrated that the Arg undergoes the greatest conformational change induced by CL interaction. In addition, Pletneva’s group94 provided a new biophysical evidence for Groves’ hypothesis in 2013. In this research, they created seven different Dns-labelled horse heart cyt c variants as well as a fluorescent liposome system to study the detailed molecular mechanism and kinetics of the cyt c unfolding process. They provided a picture of the interaction sites of cyt c with the membrane and the subsequent conformational change at the amino acid level of resolution. Following the interaction with the membrane, the breaking of the hydrogen bond between Pro 44 and His 26 is a key determinant that loosens the Met 80-containing loop, which has been known as a key feature of the folding.48 This process results in two effects: the breaking of the Met 80–heme interaction to increase the peroxidase activity, and the collapse of the overall structure which further leads to the two helical segments partially implanting into the membrane, forming the extended conformation. To summarize, this research provided a clearer explanation of both of the observed phenomena.
Some other important experimental data also stand out to demonstrate the possibility of this hypothesis. Cell penetrating peptides (CPPs) are a group of peptides with a strong ability and tendency to pass through the cellular membrane simultaneously. Howl’s group95 found that the amino acid sequences from 77–101 and 86–101 of cyt c are two potential CPPs, and 77–101 is extremely powerful. In their CPP study, a novel prediction algorithm, QSAR, was used to analyze and identify CPPs from human cyt c. Next, they quantitatively tested a series of cyt c C-terminal helical segments with different lengths. The results showed that cyt c 77–101, together with its derivatives, were all efficient CPPs, which agreed with the apoptotic induction feature of cyt c.
Finally, Groves’ theory can be a potential solution to some long-existing puzzles. For example, Kagan and co-workers56 also observed that most but not all cyt c proteins can permeate the mitochondria in the presence of alamethicin. If the CL oxidant can break the membrane, all cyt c proteins can theoretically leak. They assumed that the 15% cyt c inside might be in a CL-bound form. This observation agrees with Groves’ theory: CL-bound Cyt c might be a membrane-binding oligomer involved in pore formation, which is unable to leak due to its role in pore maintenance.
According to all the evidences above, Groves et al. have provided a novel and visible hypothesis from a different angle, that the extended form of cyt c is sufficient to induce membrane leakage independently from its peroxidase activity or membrane formation proteins. In this case, the membrane breaking process can be the result of two parallel pathways: conformational change, which (1) may or (2) may not lead to the oxidation of CL. The cyt c–CL interaction plays a role in both pathways.
5. Conclusions and outlooks
In addition to its well-characterized role as an electron carrier, cyt c has attracted increasing interest recently, because of its important role in the pre-apoptotic process. Ironically, cyt c undertakes two opposite tasks: under physiological conditions, cyt c plays an irreplaceable transporter role in the mitochondria oxidative phosphorylation pathway, which is the major energy source for cells to survive. However, under pre-apoptotic conditions, cyt c can be the executioner of the cell, delivering the death signal outside of the mitochondria. At the beginning of the process, cyt c binds to negatively charged membrane lipids such as CL through strong ionic interactions,88 which have been widely verified and accepted. Compared to the interaction, subsequent steps leading to apoptosis have been much less depicted. Several different theories have been provided trying to explain the membrane leakage phenomena in the pre-apoptotic signalling chain.By combining these theories together, we can get an overview of the whole pathway. The binding of cyt c to CL results in a conformational change of cyt c to an extended form, which provides peroxidase activity, which leads to the release of cyt c from mitochondria. In this theory, the oxidation of CL might cause membrane leakage. An alternative explanation is that a loosely bound form of cyt c can pass the membrane with the help of other proteins. What is more, an extended cyt c conformer is now hypothesized to lead to membrane leaking by itself, something which needs to be further verified.
Besides the experiments in vitro, or at the organelle level in cell-free systems, we are looking forward to the study of in vivo systems to demonstrate all hypotheses. C. elegans has been shown to have cyt c–CL interactions with similar properties,68 and thus it might be a potential working system. Until now, although we have some biophysical studies on cytochrome family proteins,96 and downstream protein–protein interaction pairs,97 no crystal structure or electron microscope structure is known for both the cyt c–CL binding system and cyt c in its membrane binding form. The development of structural biology tools, such as advances in electron microscopy,98 may bring us a new window into more detailed structures, followed by mechanistic and kinetic studies in the near future. It should be mentioned that with lots of protein chemical biology tools at hand, we can study cyt c in different unnatural structures,99 or make use of newly-developed protein in vivo labelling methods,100 for further investigations.
All the information about cyt c-induced apoptosis together suggests that the cyt c–CL complex might be a potential target for evaluation and further anti-apoptotic drug design.54 Specifically, we propose that the extended form of membrane-inserted cyt c might be a promising target, since the targets for more than 1/2 of current therapeutic drugs are membrane proteins.101
Acknowledgements
This work was supported by the National Natural Science Foundation of China (21372058 to Y. M. L.). We also thank Prof. John T. Groves, Dr Yang Lv and Christin Monoor in Princeton University for discussion and suggestions.Notes and references
- R. A. Scott and A. G. Mauk, Cytochrome c: a multidisciplinary approach, Univ Science Books, 1996 Search PubMed.
- G. Pettigrew and G. Moore, Cytochromes c Biological Aspects, Springer, Berlin, 1990 Search PubMed.
- C. R. Hackenbrock, B. Chazotte and S. S. Gupte, J. Bioenerg. Biomembr., 1986, 18, 331–368 CrossRef CAS.
- L. Banci, I. Bertini, H. B. Gray, C. Luchinat, T. Reddig, A. Rosato and P. Turano, Biochemistry, 1997, 36, 9867–9877 CrossRef CAS PubMed.
- H. Maity, M. Maity and S. Walter Englander, J. Mol. Biol., 2004, 343, 223–233 CrossRef CAS PubMed.
- J. R. Winkler, Curr. Opin. Chem. Biol., 2004, 8, 169–174 CrossRef CAS PubMed.
- A. Stivala, M. Wybrow, A. Wirth, J. C. Whisstock and P. J. Stuckey, Bioinformatics, 2011, 27, 3315–3316 CrossRef CAS PubMed.
- Y.-L. P. Ow, D. R. Green, Z. Hao and T. W. Mak, Nat. Rev. Mol. Cell Biol., 2008, 9, 532–542 CrossRef CAS PubMed.
- S. J. Riedl and G. S. Salvesen, Nat. Rev. Mol. Cell Biol., 2007, 8, 405–413 CrossRef CAS PubMed.
- X. Liu, C. N. Kim, J. Yang, R. Jemmerson and X. Wang, Cell, 1996, 86, 147–157 CrossRef CAS.
- K. Li, Y. Li, J. M. Shelton, J. A. Richardson, E. Spencer, Z. J. Chen, X. Wang and R. S. Williams, Cell, 2000, 101, 389–399 CrossRef CAS.
- H. Zou, Y. Li, X. Liu and X. Wang, J. Biol. Chem., 1999, 274, 11549–11556 CrossRef CAS PubMed.
- H. Bayir, B. Fadeel, M. Palladino, E. Witasp, I. Kurnikov, Y. Tyurina, V. Tyurin, A. Amoscato, J. Jiang, P. Kochanek, S. DeKosky, J. Greenberger, A. Shvedova and V. Kagan, Biochim. Biophys. Acta, Bioenerg., 2006, 1757, 648–659 CrossRef CAS PubMed.
- W. Martin, Proc. R. Soc. London, Ser. B, 1999, 266, 1387–1395 CrossRef.
- F. Sherman, J. W. Stewart, E. Margoliash, J. Parker and W. Campbell, Proc. Natl. Acad. Sci. U. S. A., 1966, 55, 1498 CrossRef CAS.
- O. F. Walasek and E. Margoliash, J. Biol. Chem., 1977, 252, 830–834 CAS.
- B. Kadenbach, Eur. J. Biochem., 1970, 12, 392–398 CrossRef CAS PubMed.
- B. Hennig and W. Neupert, Eur. J. Biochem., 1981, 121, 203–212 CrossRef CAS PubMed.
- P. Giegé, J. Grienenberger and G. Bonnard, Mitochondrion, 2008, 8, 61–73 CrossRef PubMed.
- R. S. Zitomer and D. L. Nichols, J. Bacteriol., 1978, 135, 39–44 CAS.
- Y. Feng, A. J. Wand and S. G. Sligar, Biochemistry, 1991, 30, 7711–7717 CrossRef CAS.
- J. M. Shilane, Covalent Attachment of Synthetic Porphyrins to Apocytochrome C: Toward Triggering Apoptosis in Cancer Cells, ProQuest, 2008 Search PubMed.
- M. Schlame, S. Shanske, S. Doty, T. König, T. Sculco, S. DiMauro and T. J. Blanck, J. Lipid Res., 1999, 40, 1585–1592 CAS.
- G. Daum, Biochim. Biophys. Acta, Rev. Biomembr., 1985, 822, 1–42 CrossRef CAS.
- J. Krebs, H. Hauser and E. Carafoli, J. Biol. Chem., 1979, 254, 5308–5316 CAS.
- C. Hesler, M. A. Carroll and D. Haldar, J. Biol. Chem., 1985, 260, 7452–7456 CAS.
- K. Hostetler, H. Van Den Bosch and L. Van Deenen, Biochim. Biophys. Acta, Lipids Lipid Metab., 1972, 260, 507–513 CrossRef CAS.
- M. Schlame and D. Haldar, J. Biol. Chem., 1993, 268, 74–79 CAS.
- M. Degli Esposti, I. Cristea, S. Gaskell, Y. Nakao and C. Dive, Cell Death Differ., 2003, 10, 1300–1309 CrossRef PubMed.
- D. R. Green and J. C. Reed, Science, 1998, 281, 1309–1311 CrossRef CAS.
- E. B. Divito and M. Cascio, Chem. Rev., 2013, 113, 7343–7353 CrossRef CAS PubMed.
- P. Nicholls, Biochim. Biophys. Acta, Rev. Bioenerg., 1974, 346, 261–310 CrossRef CAS.
- B. De Kruijff and P. Cullis, Biochim. Biophys. Acta, Biomembr., 1980, 602, 477–490 CrossRef CAS.
- W. E. Ford and G. Tollin, Photochem. Photobiol., 1982, 35, 809–819 CrossRef CAS PubMed.
- Y. Fang and G. Tollin, Photochem. Photobiol., 1988, 47, 751–758 CrossRef CAS PubMed.
- J. D. Cortese, A. L. Voglino and C. R. Hackenbrock, Biochim. Biophys. Acta, Bioenerg., 1995, 1228, 216–228 CrossRef.
- M. Rytömaa and P. K. Kinnunen, J. Biol. Chem., 1995, 270, 3197–3202 CrossRef PubMed.
- T. Heimburg and D. Marsh, Biophys. J., 1995, 68, 536–546 CrossRef CAS.
- T. Pinheiro, Biochimie, 1994, 76, 489–500 CrossRef CAS.
- Z. Salamon and G. Tollin, Biophys. J., 1996, 71, 848–857 CrossRef CAS.
- T. J. Pinheiro, G. A. Elöve, A. Watts and H. Roder, Biochemistry, 1997, 36, 13122–13132 CrossRef CAS PubMed.
- S. L. Iverson and S. Orrenius, Arch. Biochem. Biophys., 2004, 423, 37–46 CrossRef CAS PubMed.
- R. Demel, W. Jordi, H. Lambrechts, H. Van Damme, R. Hovius and B. De Kruijff, J. Biol. Chem., 1989, 264, 3988–3997 CAS.
- F. L. Hoch, Biochim. Biophys. Acta, Rev. Biomembr., 1992, 1113, 71–133 CrossRef CAS.
- A. Muga, H. H. Mantsch and W. K. Surewicz, Biochemistry, 1991, 30, 7219–7224 CrossRef CAS.
- M. M. Krishna, Y. Lin, J. N. Rumbley and S. Walter Englander, J. Mol. Biol., 2003, 331, 29–36 CrossRef CAS.
- J. F. Leszczynski and G. D. Rose, Science, 1986, 234, 849–855 CAS.
- F. Sinibaldi, B. D. Howes, M. C. Piro, P. Caroppi, G. Mei, F. Ascoli, G. Smulevich and R. Santucci, JBIC, J. Biol. Inorg. Chem., 2006, 11, 52–62 CrossRef CAS PubMed.
- M. M. Krishna, Y. Lin, L. Mayne and S. Walter Englander, J. Mol. Biol., 2003, 334, 501–513 CrossRef CAS PubMed.
- G. Balakrishnan, Y. Hu, O. F. Oyerinde, J. Su, J. T. Groves and T. G. Spiro, J. Am. Chem. Soc., 2007, 129, 504–505 CrossRef CAS PubMed.
- R. Jemmerson, J. Liu, D. Hausauer, K.-P. Lam, A. Mondino and R. D. Nelson, Biochemistry, 1999, 38, 3599–3609 CrossRef CAS PubMed.
- I. L. Nantes, M. R. Zucchi, O. R. Nascimento and A. Faljoni-Alario, J. Biol. Chem., 2001, 276, 153–158 CrossRef CAS PubMed.
- S. Oellerich, S. Lecomte, M. Paternostre, T. Heimburg and P. Hildebrandt, J. Phys. Chem. B, 2004, 108, 3871–3878 CrossRef CAS.
- V. E. Kagan, H. A. Bayır, N. A. Belikova, O. Kapralov, Y. Y. Tyurina, V. A. Tyurin, J. Jiang, D. A. Stoyanovsky, P. Wipf and P. M. Kochanek, Free Radical Biol. Med., 2009, 46, 1439–1453 CrossRef CAS PubMed.
- Y. Nakagawa, Ann. N. Y. Acad. Sci., 2004, 1011, 177–184 CrossRef CAS PubMed.
- V. E. Kagan, V. A. Tyurin, J. Jiang, Y. Y. Tyurina, V. B. Ritov, A. A. Amoscato, A. N. Osipov, N. A. Belikova, A. A. Kapralov and V. Kini, Nat. Chem. Biol., 2005, 1, 223–232 CrossRef CAS PubMed.
- N. A. Belikova, Y. A. Vladimirov, A. N. Osipov, A. A. Kapralov, V. A. Tyurin, M. V. Potapovich, L. V. Basova, J. Peterson, I. V. Kurnikov and V. E. Kagan, Biochemistry, 2006, 45, 4998–5009 CrossRef CAS PubMed.
- V. A. Tyurin, Y. Y. Tyurina, W. Feng, A. Mnuskin, J. Jiang, M. Tang, X. Zhang, Q. Zhao, P. M. Kochanek and R. S. Clark, J. Neurochem., 2008, 107, 1614–1633 CrossRef CAS PubMed.
- Y. Y. Tyurina, V. A. Tyurin, M. W. Epperly, J. S. Greenberger and V. E. Kagan, Free Radical Biol. Med., 2008, 44, 299–314 CrossRef CAS PubMed.
- V. Kagan, Y. Tyurina, H. Bayir, C. Chu, A. Kapralov, I. Vlasova, N. Belikova, V. Tyurin, A. Amoscato and M. Epperly, Chem.-Biol. Interact., 2006, 163, 15–28 CrossRef CAS PubMed.
- M. Ott, J. D. Robertson, V. Gogvadze, B. Zhivotovsky and S. Orrenius, Proc. Natl. Acad. Sci. U. S. A., 2002, 99, 1259–1263 CrossRef CAS PubMed.
- E. V. Pletneva, Z. Zhao, T. Kimura, K. V. Petrova, H. B. Gray and J. R. Winkler, J. Inorg. Biochem., 2007, 101, 1768–1775 CrossRef CAS PubMed.
- E. V. Pletneva, H. B. Gray and J. R. Winkler, Proc. Natl. Acad. Sci. U. S. A., 2005, 102, 18397–18402 CrossRef CAS PubMed.
- E. V. Pletneva, H. B. Gray and J. R. Winkler, J. Am. Chem. Soc., 2005, 127, 15370–15371 CrossRef CAS PubMed.
- J. Hanske, J. R. Toffey, A. M. Morenz, A. J. Bonilla, K. H. Schiavoni and E. V. Pletneva, Proc. Natl. Acad. Sci. U. S. A., 2012, 109, 125–130 CrossRef CAS PubMed.
- Y. Hong, J. Muenzner, S. K. Grimm and E. V. Pletneva, J. Am. Chem. Soc., 2012, 134, 18713–18723 CrossRef CAS PubMed.
- H. Neuweiler, C. M. Johnson and A. R. Fersht, Proc. Natl. Acad. Sci. U. S. A., 2009, 106, 18569–18574 CrossRef CAS PubMed.
- A. J. Vincelli, D. S. Pottinger, F. Zhong, J. Hanske, S. P. G. Rolland, B. Conradt and E. V. Pletneva, Biochemistry, 2013, 52, 653–666 CrossRef CAS PubMed.
- S. Brenner, ChemBioChem, 2003, 4, 683–687 CrossRef CAS PubMed.
- S. Hirota, Y. Hattori, S. Nagao, M. Taketa, H. Komori, H. Kamikubo, Z. Wang, I. Takahashi, S. Negi and Y. Sugiura, Proc. Natl. Acad. Sci. U. S. A., 2010, 107, 12854–12859 CrossRef CAS PubMed.
- Y. Hatefi, Eur. J. Biochem., 1993, 218, 759–767 CrossRef CAS PubMed.
- H. Imamura, K. P. H. Nhat, H. Togawa, K. Saito, R. Iino, Y. Kato-Yamada, T. Nagai and H. Noji, Proc. Natl. Acad. Sci. U. S. A., 2009, 106, 15651–15656 CrossRef CAS PubMed.
- D. B. Craig and C. J. Wallace, Biochemistry, 1995, 34, 2686–2693 CrossRef CAS.
- D. Genini, I. Budihardjo, W. Plunkett, X. Wang, C. J. Carrera, H. B. Cottam, D. A. Carson and L. M. Leoni, J. Biol. Chem., 2000, 275, 29–34 CrossRef CAS PubMed.
- L. M. Zheng, A. Zychlinsky, C.-C. Liu, D. M. Ojcius and J. Young, J. Cell Biol., 1991, 112, 279–288 CrossRef CAS.
- T. Noguchi, K. Ishii, H. Fukutomi, I. Naguro, A. Matsuzawa, K. Takeda and H. Ichijo, J. Biol. Chem., 2008, 283, 7657–7665 CrossRef CAS PubMed.
- E. K. Tuominen, K. Zhu, C. J. Wallace, I. Clark-Lewis, D. B. Craig, M. Rytömaa and P. K. Kinnunen, J. Biol. Chem., 2001, 276, 19356–19362 CrossRef CAS PubMed.
- J.-B. Denault and G. S. Salvesen, Apoptosis and Cancer, Springer, 2008, pp. 191–220 Search PubMed.
- A. Patriarca, T. Eliseo, F. Sinibaldi, M. C. Piro, R. Melis, M. Paci, D. O. Cicero, F. Polticelli, R. Santucci and L. Fiorucci, Biochemistry, 2009, 48, 3279–3287 CrossRef CAS PubMed.
- F. Sinibaldi, E. Droghetti, F. Polticelli, M. C. Piro, D. Di Pierro, T. Ferri, G. Smulevich and R. Santucci, J. Inorg. Biochem., 2011, 105, 1365–1372 CrossRef CAS PubMed.
- F. Sinibaldi, G. Mei, F. Polticelli, M. C. Piro, B. D. Howes, G. Smulevich, R. Santucci, F. Ascoli and L. Fiorucci, Protein Sci., 2005, 14, 1049–1058 CrossRef CAS PubMed.
- E. J. Snider, J. Muenzner, J. R. Toffey, Y. Hong and E. V. Pletneva, Biochemistry, 2013, 52, 993–995 CrossRef CAS PubMed.
- H. Yu, I. Lee, A. R. Salomon, K. Yu and M. Hüttemann, Biochim. Biophys. Acta, Bioenerg., 2008, 1777, 1066–1071 CrossRef CAS PubMed.
- I. Lee, A. R. Salomon, K. Yu, J. W. Doan, L. I. Grossman and M. Hüttemann, Biochemistry, 2006, 45, 9121–9128 CrossRef CAS PubMed.
- P. J. Spooner and A. Watts, Biochemistry, 1991, 30, 3871–3879 CrossRef CAS.
- T. Heimburg and D. Marsh, Biophys. J., 1993, 65, 2408–2417 CrossRef CAS.
- H. Fazelinia, M. Xu, H. Cheng and H. Roder, J. Am. Chem. Soc., 2014, 136, 733–740 CrossRef CAS PubMed.
- L. Brown and K. Wüthrich, Biochim. Biophys. Acta, Biomembr., 1977, 468, 389–410 CrossRef CAS.
- Y. Tamba and M. Yamazaki, Biochemistry, 2005, 44, 15823–15833 CrossRef CAS PubMed.
- A. Moscho, O. Orwar, D. T. Chiu, B. P. Modi and R. N. Zare, Proc. Natl. Acad. Sci. U. S. A., 1996, 93, 11443–11447 CrossRef CAS.
- P. A. Beales, C. L. Bergstrom, N. Geerts, J. T. Groves and T. K. Vanderlick, Langmuir, 2011, 27, 6107 CrossRef CAS PubMed.
- C. L. Bergstrom, P. A. Beales, Y. Lv, T. K. Vanderlick and J. T. Groves, Proc. Natl. Acad. Sci. U. S. A., 2013, 110, 6269–6274 CrossRef PubMed.
- B. S. Rajagopal, G. G. Silkstone, P. Nicholls, M. T. Wilson and J. A. Worrall, Biochim. Biophys. Acta, Bioenerg., 2012, 1817, 780–791 CrossRef CAS PubMed.
- J. Muenzner, J. R. Toffey, Y. Hong and E. V. Pletneva, J. Phys. Chem. B, 2013, 117, 12878–12886 CrossRef CAS PubMed.
- S. Jones, T. Holm, I. Mäger, Ü. Langel and J. Howl, Chem. Biol., 2010, 17, 735–744 CrossRef CAS PubMed.
- C. Hunte, J. Koepke, C. Lange, T. Roßmanith and H. Michel, Structure, 2000, 8, 669–684 CrossRef CAS.
- Y. Shi, Nat. Struct. Biol., 2001, 8, 394–401 CrossRef CAS PubMed.
- A. W. P. Fitzpatrick, U. J. Lorenz, G. M. Vanacore and A. H. Zewail, J. Am. Chem. Soc., 2013, 135, 19123–19126 CrossRef CAS PubMed.
- J. S. Zheng, S. Tang, Y.-K. Qi, Z. P. Wang and L. Liu, Nat. Protoc., 2013, 8, 2483–2495 CrossRef CAS PubMed.
- Z. Wang, X. Ding, S. Li, J. Shi and Y. Li, RSC Adv., 2014, 4, 7235–7245 RSC.
- M. A. Yıldırım, K.-I. Goh, M. E. Cusick, A.-L. Barabási and M. Vidal, Nat. Biotechnol., 2007, 25, 1119 CrossRef PubMed.
| This journal is © The Royal Society of Chemistry 2015 |