
Catalytic oxidation and adsorption of elemental mercury over nanostructured CeO2–MnOx catalyst†
Deshetti Jampaiahab,
Katie M. Turb,
Perala Venkataswamya,
Samuel J. Ippolitob,
Ylias M. Sabrib,
James Tardiob,
Suresh K. Bhargava*b and
Benjaram M. Reddy*a
aRMIT-IICT Joint Research Centre, CSIR-Indian Institute of Chemical Technology, Uppal Road, Hyderabad-500 007, India. E-mail: bmreddy@iict.res.in; mreddyb@yahoo.com; Fax: +91 40 2716 0921; Tel: +91 40 2719 3510
bCentre for Advanced Materials & Industrial Chemistry (CAMIC), School of Applied Sciences, RMIT University, GPO BOX 2476, Melbourne-3001, Australia. E-mail: suresh.bhargava@rmit.edu.in; Tel: +61 3 9925 3365
First published on 23rd March 2015
Abstract
A nanostructured CeO2–MnOx catalyst was synthesized by a coprecipitation method and subjected to different calcination temperatures at 773 and 1073 K to understand the surface structure and the thermal stability. The structural and redox properties were deeply investigated by various techniques, namely, X-ray diffraction (XRD), inductively coupled plasma-optical emission spectroscopy (ICP-OES), Brunauer–Emmett–Teller (BET) surface area, transmission electron microscopy (TEM), Raman spectroscopy (RS), hydrogen-temperature programmed reduction (H2-TPR), and X-ray photoelectron spectroscopy (XPS). The CeO2–MnOx catalyst calcined at 773 K was tested towards elemental mercury (Hg0) oxidation and the achieved results are compared with the pure CeO2 and MnOx. The XRD and TEM results confirmed the incorporation of Mn ions into the ceria lattice and the formation of a nanostructured solid solution, respectively. The RS and TPR results showed that the CeO2–MnOx catalyst exhibits more oxygen vacancies with superior redox ability over CeO2 and MnOx. XPS analysis indicates that Ce and Mn existed in multiple oxidation states. Compared to pure CeO2 and MnOx, the CeO2–MnOx catalyst exhibited greater Hg0 oxidation efficiency (Eoxi) of 11.7, 33.5, and 89.6% in the presence of HCl, O2, and HCl/O2-mix conditions, respectively. The results clearly indicated that the HCl/O2-mix had a promotional effect on the catalytic Hg0 oxidation. This was most likely due to the presence of surface oxygen species and oxygen vacancies being generated by a synergetic effect between CeO2 and MnOx. In the presence of HCl, the CeO2–MnOx catalyst exhibited good adsorption efficiency (Eads) of 92.4% over pure CeO2 (46.5%) and MnOx (80.6%). It was found that increasing the operating temperature from 423 to 573 K resulted in considerable increase of Eoxi and a decrease in the sorption of Hg0 on the catalyst.
1. Introduction
The emission of mercury (Hg) from anthropogenic sources is becoming a serious global concern that has attracted considerable attention in recent years. Mercury is a volatile and persistent pollutant that accumulates in the food chain and has serious neurological health effects in humans.1,2 Among various emission sources, the coal-fired power plants account for 24% of global anthropogenic Hg emissions and they are considered as the major emission source partly due to their unintentional Hg emissions during heating and power generation activities.3 In recent decades, many countries, especially in the developed countries, have taken steps to reduce Hg emissions. Generally, in a coal derived flue gas, there are three forms of Hg, namely, elemental mercury (Hg0), oxidized mercury (Hg2+), and mercury associated with particulate matter (HgP).4 Unlike Hg2+ and Hgp, Hg0 is difficult to remove using wet flue gas desulfurization (FGD) units and electrostatic precipitators (ESP) due to its high volatility and low solubility in water.5,6 A variety of technologies have been investigated to constrain Hg0 from the flue gas. Among them, the current state-of-art technology for controlling Hg0 emissions is Activated Carbon Injection (ACI).7–9 However, this technology has a few drawbacks such as high operation expenses, poor capacity, narrow temperature range of application, and slow adsorption and regeneration rates.10 In order to overcome these concerns, the catalytic oxidation of Hg0 to Hg2+ could be a promising approach for controlling anthropogenic Hg0 emissions from coal-fired power plants in a cost effective and environmentally friendly manner.11It is well-known that coal-fired power plants emit Hg0 into the environment during the combustion processes that occur at high temperatures (up to 1700 K). Subsequently, homogeneous oxidation can take place during the post combustion phase when temperatures drop to ranging from approximately 750 to 900 K allowing the formation of HgCl2. Similarly there is a possibility of formation of oxidized Hg species (e.g. HgCl2 or HgO) by heterogeneous catalytic oxidation as the stack gas temperature drops to 398–598 K. Accordingly, Hg0 vapor is known to mostly react with Cl2, HCl, and O2 in the stack gas, and undergo reactions to a lower extent with other species (e.g. NH3, N2O, and H2S).12,13 For the oxidization of elemental mercury by chlorine and oxygen, there is also a correlation between HCl concentration in the flue gas and the extent of elemental mercury oxidised.14 These facts show that the occurrence of Hg0 oxidation largely relies on the presence of oxidants such as HCl and O2. So, catalytic oxidation of Hg0 using gaseous O2 and HCl in the flue gas as the oxidants is a simple and economical method for Hg0 control. However, the major challenge is the selection of the proper catalysts for this purpose. In recent years, transition metal oxides appear to be promising candidates due to their much lower cost, high catalytic activity towards catalytic oxidation reactions and temperature stability relative to noble metal based catalysts.15 Several metal oxides such as CuO, Cu2O, MnOx, Fe2O3, TiO2, VOx/TiO2, and Co/TiO2 have already been investigated for the catalytic Hg0 oxidation reaction (Hg0 → Hg2+).16–18 These studies have demonstrated the feasibility of using metal oxides to catalyze the oxidation of Hg0 in the presence of hydrochloric acid (HCl) according to the following reaction:
| Hg0 + 2HCl + 1/2O2 → HgCl2 + H2O | (1) |
Among the various investigated metal oxide catalysts, manganese oxides have been studied extensively for several catalytic oxidation reactions such as partial or total oxidation of hydrocarbons due to its ability to exist in multiple oxidation states.19 In terms of catalyst efficiency, it is well reported that the Hg0 oxidation efficiency over MnOx/alumina and modified MnOx/alumina catalysts can reach more than 90%, and Mn4+ species are the most active components among the various manganese valence states.20 Ji et al.21 also reported that MnOx/TiO2 catalyst is effective for Hg0 oxidation with high capacity (17.4 mg Hg0 g−1 catalyst) at elevated temperatures of 448–473 K. The observed elemental mercury capacities were much higher than the capacities of commercially available activated carbons. They believed that Hg0 oxidation over Mn-based catalysts could be explained by Mars-Massen mechanism because HgO was found in the spent catalyst and lattice oxygen participated during the reaction. In other words, surface oxygen including lattice oxygen, chemisorbed or weakly bonded oxygen are very important for Hg0 oxidation over MnOx based catalysts.22 Although, MnOx materials showed excellent catalytic performance for Hg0 conversion at higher temperatures, they were less active at low temperatures with the presence of SO2 adding a significantly negative effect on Hg0 conversion.23,24 Therefore, a combination of MnOx and a material with a large oxygen storage capacity (OSC) is thought to facilitate Hg0 oxidation at lower temperatures around 423 K. An example is the CeO2 catalyst which has been extensively studied for various catalytic processes due to its high OSC as a result of its property of storing and releasing O2 via the Ce4+/Ce3+ redox couple.25 The success of CeO2 stems from the easy generation of oxygen vacancies that facilitate activation and transport of oxygen species. The generated surface oxygen vacancies and bulk oxygen species with relatively high mobility are very active for oxidation processes.5 Recently, various supports such as modified pillared clays (PILCs)-, titania (TiO2)-, and alumina (Al2O3)- supported CeO2–MnOx mixed oxides were studied for Hg0 oxidation.20,25,26 It was found that the synergy between CeO2 and MnOx as well as facile oxygen mobility were responsible for superior Hg0 oxidation performance. To the authors' knowledge, the unsupported CeO2–MnOx mixed oxide for mercury oxidation rarely reported in the literature. Hence, the major objective of the present study was to investigate Hg0 oxidation and adsorption efficiencies using CeO2–MnOx as the catalyst under different industrial flue gas conditions.
2. Experimental
2.1. Catalyst preparation
The CeO2–MnOx (Ce/Mn 7![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) :3 mole ratio based on oxides) catalyst was prepared by a coprecipitation method using Ce(NO3)3·6H2O (Aldrich, AR grade) and Mn(NO3)2·4H2O (Merck, AR grade) precursors. In a typical procedure, requisite quantities of precursors were dissolved separately in deionized water and mixed together. After, the aqueous NH3 was added drop-wise with vigorous stirring until the precipitation was complete (pH ∼ 9). The resulting product was filtered off, washed several times with deionized water, and oven dried at 383 K for 12 h. Finally, the catalyst was calcined at 773 K for 5 h in air atmosphere and denoted as CeMnO-773. In order to investigate the thermal stability of the catalyst, some portions of the catalyst was once again heated at 1073 K for 5 h and the final catalyst was designated as CeMnO-1073. For comparison, pure ceria (CeO2) and manganese oxide (MnOx) were also prepared by the same precipitation method.
:3 mole ratio based on oxides) catalyst was prepared by a coprecipitation method using Ce(NO3)3·6H2O (Aldrich, AR grade) and Mn(NO3)2·4H2O (Merck, AR grade) precursors. In a typical procedure, requisite quantities of precursors were dissolved separately in deionized water and mixed together. After, the aqueous NH3 was added drop-wise with vigorous stirring until the precipitation was complete (pH ∼ 9). The resulting product was filtered off, washed several times with deionized water, and oven dried at 383 K for 12 h. Finally, the catalyst was calcined at 773 K for 5 h in air atmosphere and denoted as CeMnO-773. In order to investigate the thermal stability of the catalyst, some portions of the catalyst was once again heated at 1073 K for 5 h and the final catalyst was designated as CeMnO-1073. For comparison, pure ceria (CeO2) and manganese oxide (MnOx) were also prepared by the same precipitation method.
2.2. Catalyst characterization
The X-ray diffraction measurements were performed on a Rigaku Multiflex diffractometer equipped with a nickel-filtered Cu-Kα (1.5418 Å) radiation source and a scintillation counter detector. The diffraction patterns were recorded over a 2θ range of 10–80° with a 0.021 step size and using a counting time of 1 s per point. The XRD phases present in the samples were identified with the help of the Powder Diffraction File from the International Centre for Diffraction Data (PDF-ICDD). The mean crystallite size (D) was measured by applying the Scherrer equation. The cell parameters (a) of various catalysts were calculated by a standard cubic indexation method using the intensity of the base peak (111). The chemical analysis of the prepared samples was performed using inductively coupled plasma-optical emission spectroscopy (ICP-OES, Thermo Jarrel Ash model IRIS Intrepid II XDL, USA) to confirm the respective concentrations of elements in the system. For ICP analysis, approximately 50 mg of the sample was dissolved in a solution of 25 mL aqua regia and 475 mL distilled water. Then 10 mL of the above solution was diluted to 250 mL.The surface areas and pore size distribution of the as-prepared samples were determined by N2 adsorption–desorption isotherms at liquid N2 temperature (77 K) on a Micromeritics (ASAP 2000) analyzer. Specific surface area and pore size distribution were calculated by Brunauer–Emmett–Teller (BET) and Barrett–Joyner–Halenda (BJH) methods, respectively. Prior to analysis, the samples were degassed at 393 K for 2 h to remove the surface adsorbed residual moisture.
Transmission electron microscopy (TEM) studies were carried out on a JEM-2010 (JEOL) instrument equipped with a slow-scan CCD camera and at an accelerating voltage of 200 kV. Samples for TEM analysis were prepared by crushing the materials in an agate mortar and dispersing them ultrasonically in ethyl alcohol. Afterward, a drop of the dilute suspension was placed on a perforated-carbon-coated copper grid and allowed to dry by evaporation at ambient temperature.
Raman spectra were collected on a DILORXY spectrometer equipped with a liquid nitrogen-cooled CCD detector. The samples were excited with the emission line at 632 nm from an Ar+ ion laser (Spectra Physics), which was focused on the sample under the microscope with the diameter of the analyzed spot being ∼1 μm. The acquisition time was adjusted according to the intensity of the Raman scattering. The wavenumber values obtained from the spectra were precise to within 2 cm−1.
The reducibility of the catalysts was studied by H2-TPR analysis using a thermal conductivity detector of a gas chromatograph (Shimadzu). Prior to the reduction, approximately 30 mg of the sample was loaded in an isothermal zone of the reactor and pre-treated in a helium gas flow at 473 K and then cooled to room temperature. Then, the sample was heated at a rate of 10 K min−1 from ambient temperature to 1100 K in a 20 mL min−1 flow of 5% H2 in Ar. The hydrogen consumption during the reduction process was estimated by passing the effluent gas through a molecular sieve trap to remove the produced water and it was analyzed by gas chromatography using the thermal conductivity detector.
The XPS measurements were performed on a Shimadzu ESCA 3400 spectrometer using Mg-Kα (1253.6 eV) radiation as the excitation source at room temperature. The samples were maintained in a strict vacuum typically on the order of less than 10−8 Pa to avoid a large amount of noise in the spectra from contaminants. The obtained binding energies were corrected by referencing the spectra to the carbon (C 1s) peak at 284.6 eV.
2.3. Catalyst activity studies
The catalytic oxidation of Hg0 was evaluated using a bench-scale experimental system as shown in Fig. S1 (ESI†). The apparatus consisted of several components including a temperature-controlled quartz reactor, mercury speciation trapping system, HCl vapour and Hg0 vapor generators. The Hg0 oxidation tests were performed with the total gas flow rate set at 0.2 L min−1. A mercury permeation device (VICI, Metronics Inc.) was used as the source of Hg0 (∼320 μg m−3). The N2 was used as the carrier gas to transport Hg0 vapor out of the permeation tube holder. All the individual flue gases were precisely controlled by mass flow controllers. The typical composition of a simulated flue gas (SFG) mixture was 3% O2, 5% CO2, 10 ppm HCl, and balanced with dry N2. The reactor was loaded with a small portion of quartz wool to support the catalyst layer and avoid its loss. In each test, 0.4 g of catalyst was loaded into the isothermal zone of the reactor. The reactor system was gradually heated under N2 gas to reach the desired temperature (423 K). During the experiment, the catalyst was exposed to simulated flue gas for 16 h. The outlet gas stream was passed through a series of seven impingers to capture and speciate the outlet mercury. In this work, the arrangement of the traps was based on a variant of the Ontario Hydro Method (OHM),27 in which the flue gas is passed through an absorbing media (KCl and KMnO4 solutions). Then, the samples of the absorbing media are quantitatively analyzed for their Hg contents using an analytical instrument such as Agilent 7700 Series inductively coupled plasma mass spectrometer (ICP-MS). The KCl impinger solutions were employed to capture the Hg2+, whereas KMnO4 containing impinger solutions were used to capture the non-oxidised portion (i.e. elemental mercury). According to OHM, the oxidised mercury is calculated as the sum of mercury measured in the KCl impinger solutions, while the elemental mercury is the sum of the mercury measured in the KMnO4 impinger solutions. The adsorbed mercury was then determined by digesting the spent catalyst. In this procedure, a known amount of sample was combined with 1 mL of aqua regia and one drop of KMnO4 and then aged for overnight at room temperature. The purpose of the KMnO4 in the digest media was to absorb any Hg0 evolved in the closed container due to the heat released during the digestion process. Thus the digestion process dissolves all the adsorbed mercury into the supernatant, allowing the total amount of mercury to be determined using ICP-MS analysis. To establish a baseline prior to conducting the catalysis experiment, the total inlet mercury (Hg0inlet) was determined by performing a calibration experiment without the presence of any catalyst in the system. Based on this data, the Hg0 oxidation efficiency (Eoxi) and the Hg0 adsorption efficiency (Eads) of the developed catalysts were determined by using the following equations:
 | (2) |
 | (3) |
3. Results and discussion
3.1. Characterization studies
Fig. 1 illustrates the XRD patterns of MnOx, CeO2, and CeO2–MnOx mixed oxides calcined at different temperatures. The XRD pattern of MnOx calcined at 773 K represents the Mn2O3 phase, which is well matched with the cubic Mn2O3 (JCPDS card no. 01-1061).28 On the other hand, the CeO2–MnOx catalyst calcined at 773 K exhibits the characteristic peaks related to the fluorite structured CeO2 (PDF-ICDD 34-0394).29 The absence of Mn-oxide phases (MnO2, Mn2O3, MnO, and Mn3O4) has been attributed to either the dispersion of MnOx phases in ceria lattice or the Mn cations may be substituted instead of some Ce sites in the fluorite structure. Surprisingly, the CeO2–MnOx catalyst calcined at 1073 K showed very weak Mn2O3 peaks along with CeO2 peaks, which might be due to Mn-oxide phase segregation at elevated thermal treatments.29 Further, an increase in the intensity and decrease in the peak width were observed in the XRD patterns at higher calcination temperatures. This may be due to the growing degree of the crystallinity of the samples.30 The crystallite sizes were determined by using Scherrer equation for the CeO2 (111) plane, the results of which are listed in Table 1. The CeMnO-773 catalyst exhibited a lower crystallite size (7.2 nm) compared to pure CeO2 (8.9 nm). With increase in calcination temperature, the average crystallite size increased to 22.4 nm, which could be rationalized as a result of sintering.31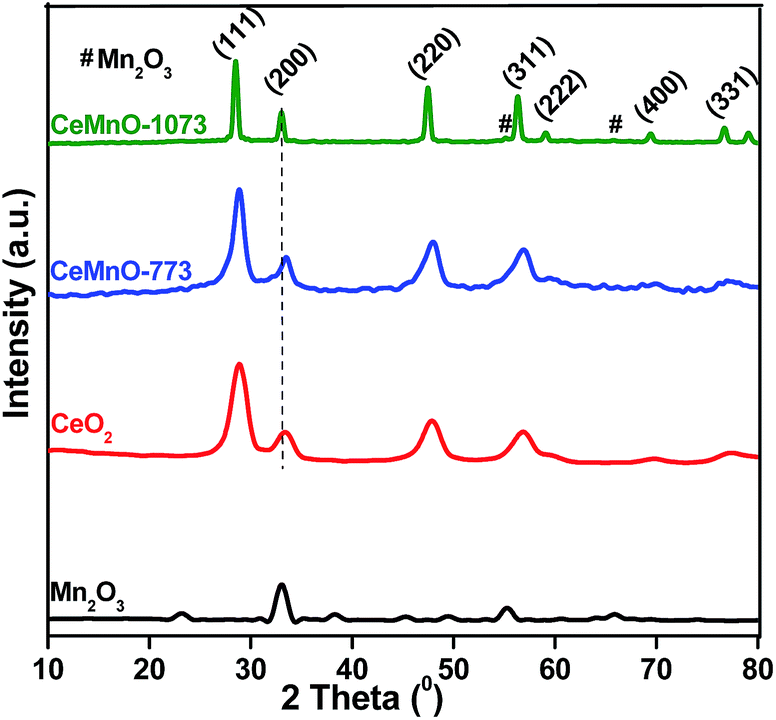 | ||
| Fig. 1 Powder XRD patterns of MnOx and CeO2 catalysts calcined at 773 K, and CeO2–MnOx catalysts calcined at 773 and 1073 K. | ||
| Catalyst | Surface area (m2 g−1) | Pore volume (cm3 g−1) | Pore size (nm) | Crystallite size (nm) | Lattice parameter (Å) |
|---|---|---|---|---|---|
| CeO2 | 39 | 0.11 | 9.8 | 8.9 | 5.41 |
| MnOx | 8 | 0.02 | 12.3 | 32.4 | 4.71 |
| CeMnO-773 | 58 | 0.44 | 6.2 | 7.2 | 5.35 |
| CeMnO-1073 | 11 | 0.18 | 7.7 | 22.4 | 5.41 |
Additionally, it was observed that the diffraction peaks of CeO2–MnOx catalysts were shifted slightly to higher angles compared to CeO2, resulting from lattice contraction due to the substitution of Ce4+ by Mnx+ in the fluorite structure.25 Because of this, the cell parameter of ceria (5.41 Å) decreases to 5.35 Å upon Mn doping (CeMnO-773 catalyst). These results imply that some manganese ions were incorporated into the ceria lattice, leading to the formation of a solid solution between manganese and cerium oxides. This observation is in agreement with Vegard's law, which holds that a linear relationship exists, at a constant temperature. As the calcination temperature increased to 1073 K, we observed a small increase in lattice parameter. Overall, these results confirm that the strong Ce–Mn–O interaction in the solid solution have effectively inhibited the crystalline growth and in turn improved the thermal stability of CeO2–MnOx catalyst.
Furthermore, the molar ratios of Ce/Mn in the CeO2–MnOx mixed oxides were determined by ICP-OES analysis, and the chemical compositions of the samples are shown in Table S1 (ESI†). The reported chemical compositions of the prepared samples were close to those of the initial mixed solutions.
Fig. 2 shows the profiles of adsorption–desorption curves of nitrogen isotherms for all the catalysts. The values of specific surface area, pore volume, and pore size of all catalysts are summarized in Table 1. The nanostructured CeMnO-773 catalyst exhibited a high BET surface area (58 m2 g−1) compared to pure CeO2 and MnOx. The high specific surface area of CeMnO-773 sample is probably due to the presence of smaller sized CeO2 crystallites. However, as the calcination temperature increases to 1073 K, the surface area decreased to 11 m2 g−1. The decline of the surface area with increasing calcination temperature could be due to the increase in the particle size with temperature. Additionally, the CeO2–MnOx catalyst calcined at 773 K exhibited lower average pore size value, while compared to pure CeO2 and MnOx. However, the average pore size increased with increasing calcination temperature due to the larger size of the CeO2 crystallites.
 | ||
| Fig. 2 Nitrogen adsorption–desorption isotherms of MnOx and CeO2 catalysts calcined at 773 K, and CeO2–MnOx catalysts calcined at 773 and 1073 K. | ||
The morphology and crystalline growth of samples were examined by TEM and HRTEM analyses and corresponding images are shown in Fig. 3. The nanosized and well dispersed particles (∼7–20 nm) can be clearly observed for both CeO2–MnOx catalysts calcined at different temperatures (Fig. 3B and C). The average particle size of CeMnO-773 catalyst was ∼7 nm, in reasonable agreement with the size calculated using the Scherrer equation (Table 1). The HRTEM images of CeMnO-773 and CeMnO-1073 samples (Fig. 3D and E) display the crystalline nature of the developed nanoparticles. The lattice fringes are clearly visible and planes with d111 spacing of 0.309 nm related to fluorite structured CeO2 were observed in both samples. It clearly indicates the thermal stability of the CeO2–MnOx catalyst.32,33
 | ||
| Fig. 3 TEM and HRTEM images of CeO2 calcined at 773 K (A), CeO2–MnOx catalysts calcined at 773 K (B and D) and 1073 K (C and E). | ||
Fig. 4 illustrates the Raman spectra of CeO2–MnOx catalysts along with pure CeO2 and MnOx. The Raman band at ∼465 cm−1 for CeO2 is related to the vibrational mode (F2g) of fluorite-type structure, which can be regarded as a symmetric breathing mode of the oxygen ions around each Ce4+ cation.34 On the other hand, the Raman spectra of CeMnO-773 and CeMnO-1073 catalysts displayed peaks at ∼453 and ∼459 cm−1, respectively. This red shift of F2g mode indicates the formation of solid solution between Ce and Mn that could deform the fluorite structure and form oxygen vacancies in the CeO2 lattice. The presence of oxygen vacancies can be confirmed by the Raman mode at ∼646 cm−1 for CeMnO-773 catalyst.35 However, for CeMnO-1073 catalyst, the peaks at ∼317, 363, and 653.9 cm−1 can be ascribed to the different phases such as Mn3O4 and Mn2O3.36 In particular, Mn2O3 phase was detected by the XRD analysis indicating that Mn2O3 was segregated from the mixed oxide at elevated temperatures, which is strongly supported by XRD results. Based on these results, it can be stated that when a Ce4+ ion is substituted with a manganese ion, an oxygen vacancy is formed to maintain the charge balance, which plays an important role for Hg0 oxidation.
 | ||
| Fig. 4 Raman spectra of MnOx and CeO2 catalysts calcined at 773 K, and CeO2–MnOx catalysts calcined at 773 and 1073 K. | ||
Fig. 5 shows the H2-TPR profiles of CeO2–MnOx catalysts along with pure CeO2 and MnOx. Generally, pure ceria exhibits two reduction peaks corresponding to the surface and bulk reduction which are centred at ∼793 and 1100 K (not shown), respectively.37 The pure MnOx also exhibited two reduction peaks at ∼603 and 745 K. The first reduction peak at ∼603 K can be ascribed to the reduction of MnO2 and/or Mn2O3 to Mn3O4 while the second peak at ∼745 K is generated because of the reduction of Mn3O4 to MnO.38 As shown from Fig. 5, the reduction peaks of the CeO2–MnOx catalysts were observed at lower temperatures than those observed for both pure CeO2 and MnOx. It can be concluded that the reduction peak at a low temperature corresponds to the reduction of manganese oxide species (MnO2 and/or Mn2O3), while the high temperature reduction peak could be attributed to the combined reductions of Mn3O4 and surface Ce-oxide species. The possible reason for this behavior is the enhancement of oxygen vacancies through the formation of solid solution, thereby increasing the lattice oxygen mobility and facilitating the diffusion of lattice oxygen from the bulk to the surface (as evidenced from Raman results).31,39 Besides, as the calcination temperature increased, the reduction peaks were observed to shift to a higher temperature compared to CeMnO-773. This may be due to the slight segregation of Mn-oxide from CeO2 lattice, which was supported by XRD and Raman results. However, the CeMnO-1073 catalyst showed prominent redox ability while compared to pure CeO2 and MnOx, indicating that the CeO2–MnOx catalyst was thermally stable. These results render that there may have formed a synergetic interaction between Ce and Mn that will possibly be useful for reactions such as the catalytic Hg0 oxidation reaction.
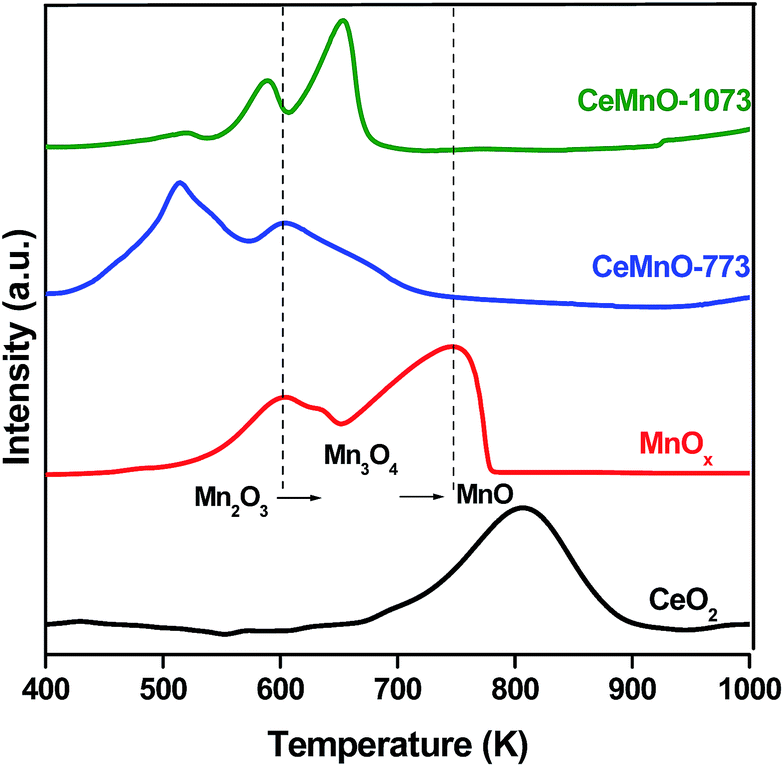 | ||
| Fig. 5 H2-TPR patterns of CeO2 and MnOx catalysts calcined at 773 K, and CeO2–MnOx catalysts calcined at 773 and 1073 K. | ||
Fig. 6A illustrates Ce 3d XP spectra of CeO2, CeMnO-773, and CeMnO-1073 samples. The main features are composed of eight peaks labelled as u and v. The peaks labelled as u are due to 3d3/2 spin–orbit states, and those labelled as v are due to the corresponding 3d5/2 states. As can be noted from Fig. 6A, the six peaks labelled as u, u′′, u′′′, v, v′′, v′′′ are featured to Ce4+ ions, and the other 2 peaks labelled as u′ and v′ are related to Ce3+ ions.40,41 From Table 2, it can be concluded that CeO2–MnOx samples exhibited the binding energy (u′′′) lower than pure ceria, indicating that the doping influences the chemical environment of ceria. It is reported that the existence of Ce3+ in CeO2 implies the formation of an oxygen vacancies.42 As can be noted from Table 3, it is clear that Ce4+ and Ce3+ coexist in the sample, however the ratio of Ce4+ to Ce3+ changes. After Mn doping into CeO2 lattice, the ratio decreased from 5.04 to 3.14, which indicates that reduced species Ce3+, together with Mn3+ played a vital role in creating charge imbalance, vacancies, and unsaturated chemical bonds on the catalyst surface.25 Therefore, the coexistence of Ce4+ and Ce3+ species at the surface could lead to the appearance of more oxygen vacancies in CeO2–MnOx catalyst, which is in agreement with the Raman analysis. Based on the observed phenomena, it can be concluded that the combination of CeO2 and MnOx could facilitate more surface oxygen for Hg0 oxidation.
 | ||
| Fig. 6 (A) Ce 3d (B) O 1s (C) Mn 2p XP spectra of fresh CeO2 and MnOx catalysts calcined at 773 K, and CeO2–MnOx catalysts calcined at 773 and 1073 K. | ||
| Catalyst | Binding energies (eV) | ||||||
|---|---|---|---|---|---|---|---|
| Ce 3d3/2 (u′′′) | Mn 2p3/2 | O 1s | |||||
| Mn4+ | Mn3+ | Mn2+ | OI | OII | OIII | ||
| MnOx | — | 643.9 | 641.6 | 640.4 | 529.3 | 531.3 | — |
| CeO2 | 916.4 | — | — | — | 530.2 | 531.9 | — |
| CeMnO-773 | 915.3 | 643.2 | 641.2 | 640.1 | 529.1 | 530.5 | 532.7 |
| CeMnO-1073 | 916.1 | 643.7 | 641.5 | 640.2 | 529.8 | 530.6 | 533.8 |
| Catalyst | Ce4+/Ce3+ | Mn4+/Mn3+ + Mn2+ | ||
|---|---|---|---|---|
| Fresh | Spent | Fresh | Spent | |
| MnOx | — | — | 0.63 | 0.25 |
| CeO2 | 5.04 | 4.87 | — | — |
| CeMnO-773 | 3.14 | 1.36 | 1.15 | 0.46 |
The O 1s spectra of pure CeO2, CeMnO-773, and CeMnO-1073 samples are shown in Fig. 6B. The O 1s spectra of pure CeO2 and MnOx indicate the presence of both chemisorbed and lattice oxygen species on the catalyst surface. On the other hand, the O 1s spectra of CeO2–MnOx catalysts contained three types of oxygen species, in which the binding energy at ∼529.1–530.2 eV was assigned to the lattice oxygen (OI) while the binding energy at ∼530.6–531.9 eV was attributed to oxygen species in the defects or hydroxyl-like groups (denoted as OII). Lastly, the binding energy at ∼532.7–533.8 eV (OIII) can be attributed to carbonates/adsorbed water.40 Interestingly, the binding energy of lattice oxygen (OI) was decreased for CeMnO-773 sample while compared to pure CeO2, which is evidenced from Table 2. It can be attributed to the O → Mn electron-transfer processes as the interactions between Ce and Mn are synergetic in nature where the dopant cation is having a decisive role towards the binding energy shift. As a result, the O → Mn in the CeO2–MnOx mixed oxide could enable the formation of very reactive electrophilic oxygen species (e.g., O2−, O−, O˙).43 It is well-known that the defect oxygen species are responsible for oxidation reactions.44 Therefore, the OII species in the CeMnO-773 sample might be helpful for Hg0 oxidation reaction.
In order to identify the oxidation states of Mn, the XPS spectra of Mn 2p was acquired and shown in Fig. 6C. It is clear that the co-existence of Mn2+, Mn3+, and Mn4+ species have been identified by the binding energies at ∼640.2, 641.5, and 643.5 eV, respectively.21,39,45 The observed binding energies of the manganese ions in the CeO2–MnOx samples are lower than pure MnO, MnO2, and Mn2O3, which might be due to a strong interaction between manganese and cerium oxides.30 As can be observed from Table 3, the CeMnO-773 sample showed more Mn4+/Mn3+ + Mn2+ ratio (1.15), while compared to pure MnOx (0.63). It can be concluded that the combination of CeO2 and MnOx results in more surface Mn4+ concentration. Several authors reported that high valance having Mn-oxides could enhance Hg0 oxidation efficiency as the Mn4+ could directly oxidize the adsorbed Hg0.13,46 Further, they explained that the presence of Mn3+ also plays an important role in Hg0 oxidation in the O2 atmosphere. Therefore, the high ratio of Mn4+/Mn3+ + Mn2+ could be responsible for better Hg0 oxidation.
3.2. Mercury oxidation
 | ||
| Fig. 7 Hg speciation over CeO2, MnOx, and CeO2–MnOx catalysts calcined at 773 K under different flue gas conditions (A) HCl (B) O2 (C) HCl and O2. | ||
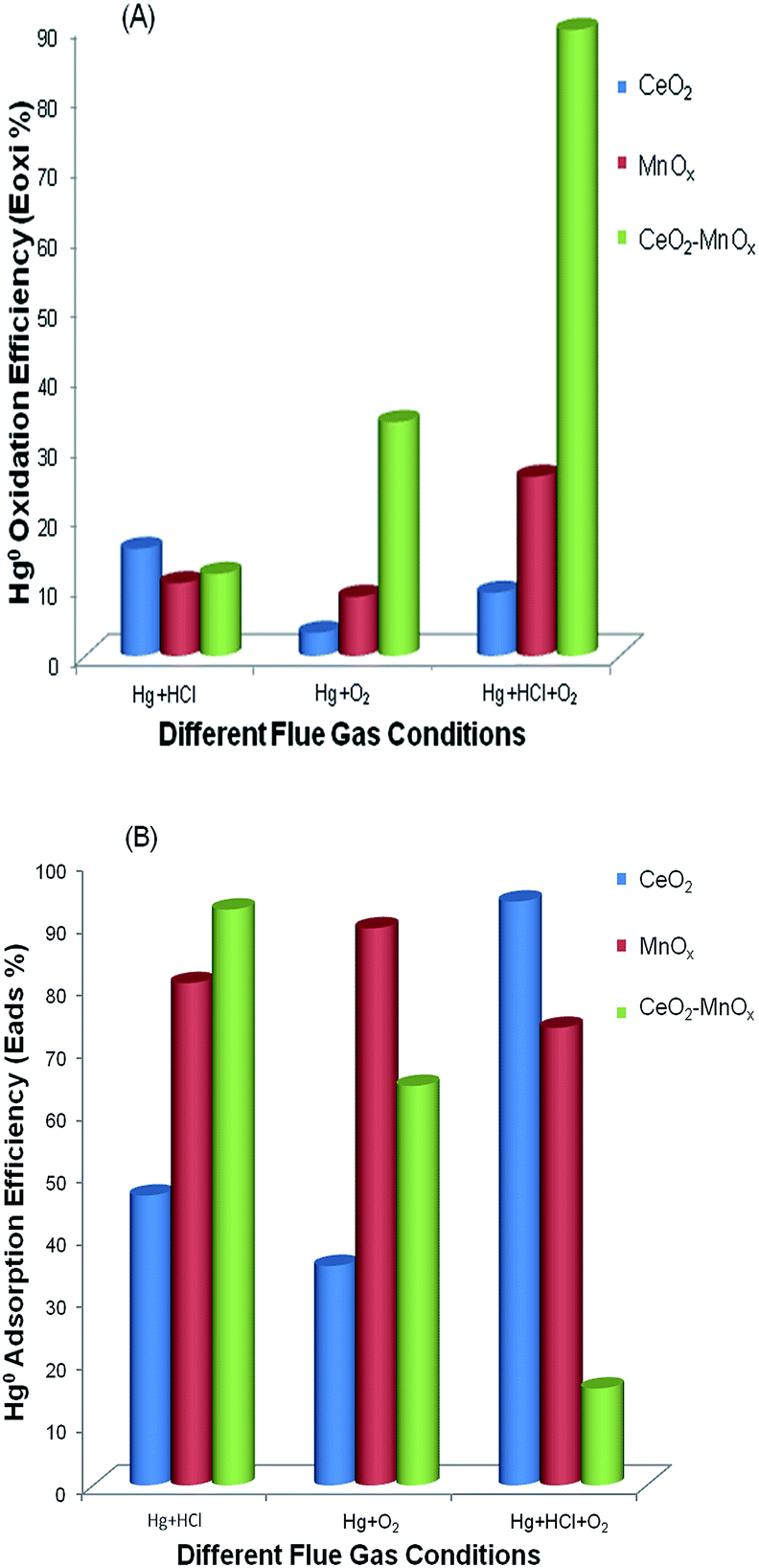 | ||
| Fig. 8 (A) Hg0 oxidation (Eoxi) and (B) Hg0 adsorption (Eads) efficiencies of CeO2, MnOx, and CeO2–MnOx catalysts calcined at 773 K under different flue gas conditions. | ||
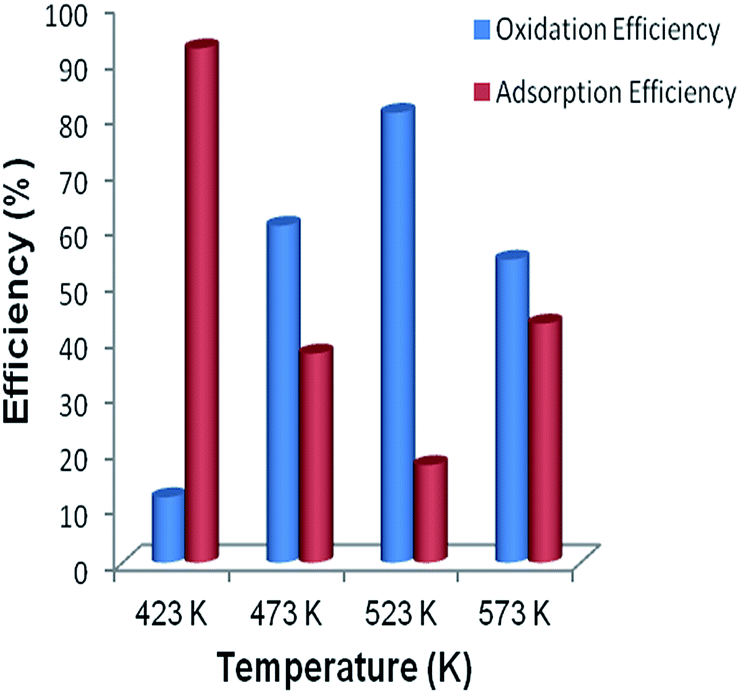 | ||
| Fig. 9 Hg0 oxidation (Eoxi) and adsorption (Eads) efficiencies of CeO2–MnOx catalysts calcined at 773 K in presence of HCl under different temperatures from 423 to 573 K. | ||
The effect of cerium to the catalysis might be attributed to its promotion on the cycle between Mn3+ and Mn4+, which would speed up the transfer of oxygen and the formation of active chlorine species. Therefore, it can be concluded that the combination of Ce and Mn in the catalyst matrix had an obvious accelerative effect on Hg0 removal relative to their individual respective oxides.
| 2HCl(g) + O* → 2Cl*(ad) + H2O | (4) |
| Hg0(g) → Hg0(ad) | (5) |
| Cl*(ad) + Hg0(ad) → HgCl*(ad) | (6) |
| HgCl*(ad) + Cl*(ad) → HgCl2(ad) | (7) |
| HgCl2(ad) → HgCl2(g) | (8) |
In this reaction, O* represents chemisorbed or lattice oxygen on the surface of CeO2–MnOx catalyst, which can be consumed by HCl. During the reaction, an intermediate product (HgCl*) is formed which subsequently oxidized by other active chlorine species to form HgCl2.50
Furthermore, to obtain more insight into the interaction between CeO2 and MnOx in the mixed oxide due to mercury adsorption in the presence of HCl and O2, XPS measurements were conducted on the spent catalysts. The Ce 3d, O 1s, and Mn 2p XP spectra of used CeO2, MnOx, and CeO2–MnOx catalysts were shown in Fig. S3A–C (ESI†), respectively. After Hg0 saturation adsorption at 423 K, the intensity of Ce4+ peaks in Ce 3d spectra decreased, which indicates a reduction of Ce4+ during Hg0 oxidation. For CeMnO-773 sample, the ratio of Ce4+/Ce3+ decreased from 3.14 to 1.36 (Table 3) after mercury oxidation and adsorption experiments. It can be due to the easily generated labile oxygen vacancies and high mobile oxygen during the redox process of Ce4+ ↔ Ce3+. In addition, this observation revealed the possibility of the reaction between cation vacancies and Hg0 during the oxidation process. As observed from Table 2 and S2 (ESI†), the significant shift of Ce 3d level of the used CeO2 catalyst to the lower energy side compared to CeO2–MnOx mixed oxide confirms the strongly electro-negative chlorine species adsorbed on CeO2 sites. On the other hand, the binding energy shifts of CeO2–MnOx are slight, due to a weak adsorption of chlorine species. Therefore, the combination of CeO2 with MnOx decreased adsorption efficiency and increased oxidation efficiency. The intensity decrease of Mn 2p peaks in the used CeO2–MnOx catalyst was due to the adsorption of mercury species on the surface. Particularly, the ratio decrease of Mn4+/Mn3+ + Mn2+ confirms the occurrence of the reaction between the active species and lattice oxygen provided by MnOx. Therefore, it could be concluded that the presence of the active Ce4+ and Mn4+ phases as well as the formation of Ce3+ (or oxygen vacancies) contributed to high catalytic activity, which are strongly supported by XPS results. The Hg 4f and Cl 2p spectra of all of the MnOx, CeO2, and CeO2–MnOx catalysts were also shown in Fig. S4A and B (ESI†), respectively. The peak at lower binding energy could be due to elemental mercury, and the peak at higher binding energy is due to either HgO or HgCl2. For MnOx sample, the binding energies of Hg 4f7/2 at 101.3 eV and Hg 4f5/2 at 103 eV correspond to the oxidized mercury (HgO).51 On the other hand, the peak due to the HgO or HgCl2 is larger for CeO2–MnOx mixed oxide compared to the other samples. It is clear that the oxidised mercury can be in the form of HgO/HgCl2. Additionally, the Cl 2p peak appearing at ∼198 eV confirm the presence of chlorine species in the form of HgCl2 on all of the used catalysts.
4. Conclusions
The nanostructured CeO2–MnOx catalyst was successfully prepared by a coprecipitation method from cerium and manganese nitrate precursors. The physicochemical properties were deeply investigated by various state-of-art techniques. The XRD and TEM results confirmed the formation of nanosized Ce0.7Mn0.3O2−δ solid solutions with a cubic phase of fluorite structure. The Raman results revealed the presence of more oxygen vacancies in the CeO2–MnOx mixed oxide compared to pure metal oxides. H2-TPR measurements show that there was a synergetic effect between CeO2 and MnOx in the mixed oxide. XPS results of fresh and spent catalysts suggested that Ce4+ and Mn4+ species contributed to more Hg0 oxidation performance. Among all the investigated catalysts, the CeO2–MnOx mixed oxide exhibited superior activity toward Hg0 oxidation compared to pure CeO2 or MnOx. The better activity could be attributed to the formation of more oxygen vacancies and facile redox behavior, which is resulted owing to strong synergetic effect between CeO2 and MnOx. The effects of various other flue gas constituents such as NOx, NH3, SO2, and water vapor on Hg0 oxidation efficiency of the developed CeO2–MnOx catalyst is the subject of future work.Acknowledgements
D. J. thanks the RMIT-IICT Joint Research Center for providing the Junior Research Fellowship.References
- J. Zhou, W. Hou, P. Qi, X. Gao, Z. Luo and K. Cen, Environ. Sci. Technol., 2013, 47, 10056–10062 CrossRef CAS PubMed.
- B. M. Reddy, N. Durgasri, T. V. Kumar and S. K. Bhargava, Catal. Rev.–Sci. Eng., 2012, 54, 344–398 CAS.
- C. Sun, C. E. Snape and H. Liu, Energy Fuels, 2013, 27, 3875–3882 CrossRef CAS.
- D. Jampaiah, K. M. Tur, S. J. Ippolito, Y. M. Sabri, J. Tardio, S. K. Bhargava and B. M. Reddy, RSC Adv., 2013, 3, 12963–12974 RSC.
- A. A. Presto, E. J. Granite, A. Karash, R. A. Hargis, W. J. O'Dow and H. W. Pennline, Energy Fuels, 2006, 20, 1941–1945 CrossRef CAS.
- S. Yang, Y. Guo, N. Yan, Z. Qu, J. Xie, C. Yang and J. Jia, J. Hazard. Mater., 2011, 186, 508–515 CrossRef CAS PubMed.
- A. P. Jones, J. W. Hoffmann, D. N. Smith, T. J. Feeley and J. T. Murphy, Environ. Sci. Technol., 2007, 41, 1365–1371 CrossRef CAS.
- A. A. Presto and E. J. Granite, Environ. Sci. Technol., 2007, 41, 6579–6584 CrossRef CAS.
- G. Luo, H. Yao, M. Xu, X. Cui, W. Chen, R. Gupta and Z. Xu, Energy Fuels, 2009, 24, 419–426 CrossRef.
- Y. M. Sabri, S. J. Ippolito and S. K. Bhargava, J. Phys. Chem. C, 2013, 117, 8269–8275 CAS.
- A. A. Presto and E. J. Granite, Environ. Sci. Technol., 2006, 40, 5601–5609 CrossRef CAS.
- K. S. Park, Y. C. Seo, S. J. Lee and J. H. Lee, Powder Technol., 2008, 180, 151–156 CrossRef CAS PubMed.
- P. Wang, S. Su, J. Xiang, F. Cao, L. Sun, S. Hu and S. Lei, Chem. Eng. J., 2013, 225, 68–75 CrossRef CAS PubMed.
- J.-R. Li, C. He, X.-S. Shang, J.-S. Chen, X.-W. Yu and Y.-J. Yao, J. Fuel Chem. Technol., 2012, 40, 241–246 CrossRef CAS.
- P. Venkataswamy, K. N. Rao, D. Jampaiah and B. M. Reddy, Appl. Catal., B, 2015, 162, 122–132 CrossRef CAS PubMed.
- Y. Gao, Z. Zhang, J. Wu, L. Duan, A. Umar, L. Sun, Z. Guo and Q. Wang, Environ. Sci. Technol., 2013, 47, 10813–10823 CrossRef CAS PubMed.
- H. Kamata, S.-I. Ueno, T. Naito, A. Yamaguchi and S. Ito, Catal. Commun., 2008, 9, 2441–2444 CrossRef CAS PubMed.
- Y. Liu, Y. Wang, H. Wang and Z. Wu, Catal. Commun., 2011, 12, 1291–1294 CrossRef CAS PubMed.
- Z. Chen, Z. Jiao, D. Pan, Z. Li, M. Wu, C.-H. Shek, C. M. L. Wu and J. K. L. Lai, Chem. Rev., 2012, 112, 3833–3855 CrossRef CAS PubMed.
- C. He, B. Shen, J. Chen and J. Cai, Environ. Sci. Technol., 2014, 48, 7891–7898 CrossRef CAS PubMed.
- L. Ji, P. M. Sreekanth, P. G. Smirniotis, S. W. Thiel and N. G. Pinto, Energy Fuels, 2008, 22, 2299–2306 CrossRef CAS.
- Q. Wan, L. Duan, K. He and J. Li, Chem. Eng. J., 2011, 170, 512–517 CrossRef CAS PubMed.
- J. Li, N. Yan, Z. Qu, S. Qiao, S. Yang, Y. Guo, P. Liu and J. Jia, Environ. Sci. Technol., 2009, 44, 426–431 CrossRef PubMed.
- Y. Guo, N. Yan, S. Yang, P. Liu, J. Wang, Z. Qu and J. Jia, J. Hazard. Mater., 2012, 213–214, 62–70 CrossRef CAS PubMed.
- H. Li, C.-Y. Wu, Y. Li and J. Zhang, Appl. Catal., B, 2012, 111–112, 381–388 CrossRef CAS PubMed.
- P. Wang, S. Su, J. Xiang, H. You, F. Cao, L. Sun, S. Hu and Y. Zhang, Chemosphere, 2014, 101, 49–54 CrossRef CAS PubMed.
- J. Q. Sun, K. S. Uhrich and R. L. Schulz, Prepr. Pap.–Am. Chem. Soc., Div. Fuel Chem., 2003, 48, 774–776 CAS.
- B. C. Sekhar, G. Babu and N. Kalaiselvi, RSC Adv., 2015, 5, 4568–4577 RSC.
- K. N. Rao, P. Bharali, G. Thrimurthulu and B. M. Reddy, Catal. Commun., 2010, 11, 863–866 CrossRef CAS PubMed.
- W.-J. Hong, S. Iwamoto, S. Hosokawa, K. Wada, H. Kanai and M. Inoue, J. Catal., 2011, 277, 208–216 CrossRef CAS PubMed.
- Z.-Q. Zou, M. Meng and Y.-Q. Zha, J. Phys. Chem. C, 2009, 114, 468–477 Search PubMed.
- H. Li, Tana, X. Zhang, X. Huang and W. Shen, Catal. Commun., 2011, 12, 1361–1365 CrossRef CAS PubMed.
- Z. Yang, Y. Zhang, W. Zhang, X. Wang, Y. Qian, X. Wen and S. Yang, J. Solid State Chem., 2006, 179, 679–684 CrossRef CAS PubMed.
- X. Li, X. Lu, Y. Meng, C. Yao and Z. Chen, J. Alloys Compd., 2013, 562, 56–63 CrossRef CAS PubMed.
- F. D. A. A. Barros, H. S. A. de Sousa, A. C. Oliveira, M. C. Junior, J. M. Filho, B. C. Viana and A. C. Oliveira, Catal. Today, 2013, 212, 127–136 CrossRef CAS PubMed.
- F. Buciuman, F. Patcas, R. Craciun and D. R. T. Zahn, Phys. Chem. Chem. Phys., 1999, 1, 185–190 RSC.
- L. Katta, G. Thrimurthulu, B. M. Reddy, M. Muhler and W. Grunert, Catal. Sci. Technol., 2011, 1, 1645–1652 CAS.
- A. Gupta, U. V. Waghmare and M. S. Hegde, Chem. Mater., 2010, 22, 5184–5198 CrossRef CAS.
- L. Qu, C. Li, G. Zeng, M. Zhang, M. Fu, J. Ma, F. Zhan and D. Luo, Chem. Eng. J., 2014, 242, 76–85 CrossRef CAS PubMed.
- P. Zhao, C. Wang, F. He and S. Liu, RSC Adv., 2014, 4, 45665–45672 RSC.
- H. Zhang, F. Gu, Q. Liu, J. Gao, L. Jia, T. Zhu, Y. Chen, Z. Zhong and F. Su, RSC Adv., 2014, 4, 14879–14889 RSC.
- Z.-Q. Zou, M. Meng and Y.-Q. Zha, J. Phys. Chem. C, 2009, 114, 468–477 Search PubMed.
- W. Xingyi, K. Qian and L. Dao, Appl. Catal., B, 2009, 86, 166–175 CrossRef PubMed.
- F. Arena, G. Trunfio, J. Negro, B. Fazio and L. Spadaro, Chem. Mater., 2007, 19, 2269–2276 CrossRef CAS.
- R. Xu, X. Wang, D. Wang, K. Zhou and Y. Li, J. Catal., 2006, 237, 426–430 CrossRef CAS PubMed.
- J. Xie, Z. Qu, N. Yan, S. Yang, W. Chen, L. Hu, W. Huang and P. Liu, J. Hazard. Mater., 2013, 261, 206–213 CrossRef CAS PubMed.
- C. Bozo, N. Guilhaume and J.-M. Herrmann, J. Catal., 2001, 203, 393–406 CrossRef CAS.
- Z.-Y. Pu, X.-S. Liu, A.-P. Jia, Y.-L. Xie, J.-Q. Lu and M.-F. Luo, J. Phys. Chem. C, 2008, 112, 15045–15051 CAS.
- P. Wang, S. Su, J. Xiang, H. You, F. Cao, L. Sun, S. Hu and Y. Zhang, Chemosphere, 2014, 101, 49–54 CrossRef CAS PubMed.
- H. Kamata, S.-i. Ueno, N. Sato and T. Naito, Fuel Process. Technol., 2009, 90, 947–951 CrossRef CAS PubMed.
- J. He, G. K. Reddy, S. W. Thiel, P. G. Smirniotis and N. G. Pinto, J. Phys. Chem. C, 2011, 115, 24300–24309 CAS.
Footnote |
| † Electronic supplementary information (ESI) available: The chemical compositions of CeO2–MnOx mixed oxides, binding energies of spent catalysts, experimental diagram, calibration experiments for Hg0inlet, Ce 3d, O 1s, Mn 2p, Hg 4f and Cl 2p XPS spectra of spent catalysts calcined at 773 K. See DOI: 10.1039/c4ra16787b |
| This journal is © The Royal Society of Chemistry 2015 |