
Enantioselective bioreductive preparation of chiral halohydrins employing two newly identified stereocomplementary reductases†
Guo-Chao Xuab,
Hui-Lei Yu*a,
Yue-Peng Shanga and
Jian-He Xu*a
aState Key Laboratory of Bioreactor Engineering, East China University of Science and Technology, and Shanghai Collaborative Innovation Center for Biomanufacturing Technology, Shanghai 200237, China. E-mail: huileiyu@ecust.edu.cn; jianhexu@ecust.edu.cn
bThe Key Laboratory of Industrial Biotechnology, Ministry of Education, School of Biotechnology, Jiangnan University, Wuxi 214122, China
First published on 9th February 2015
Abstract
Two robust stereocomplementary carbonyl reductases (DhCR and CgCR) were identified through rescreening the carbonyl reductase toolbox. Five reductases were returned through the activity and enantioselectivity assay for α-chloro-1-acetophenone and ethyl 4-chloro-3-oxo-butanate (COBE). Three reductases were stable at elevated substrate loading. Enzymatic characterization revealed that DhCR and CgCR were more thermostable. As much as 330 g COBE in 1 L biphasic reaction mixture was reduced to (S)- and (R)-3-hydroxy-4-chlorobutyrate by DhCR and CgCR (coexpressed with glucose dehydrogenase), with 92.5% and 93.0% yields, >99% ee, and total turnover numbers of 53![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) 800 and 108000, respectively. Six other α-halohydrins were asymmetrically reduced to optically pure forms at a substrate loading of 100 g L−1. Our results indicate the potential of these two stereocomplementary reductases in the synthesis of valuable α-halohydrins for pharmaceuticals.
800 and 108000, respectively. Six other α-halohydrins were asymmetrically reduced to optically pure forms at a substrate loading of 100 g L−1. Our results indicate the potential of these two stereocomplementary reductases in the synthesis of valuable α-halohydrins for pharmaceuticals.
Introduction
Asymmetric reduction of prochiral ketones is one of the most important fundamental and practical reactions for production of chiral secondary alcohols, which can be transformed into various functionalities of industrial relevance for pharmaceuticals, agrochemicals and natural products.1 Optically active α-halogenated alcohols constitute important building blocks in the synthesis of pharmaceutical and liquid crystal products.2 Most importantly, the two opposite enantiomers may have similar, different or even opposite effects. For example, ethyl (S)-3-hydroxy-4-chlorobutyrate ((S)-CHBE) is a key chiral precursor for HMG-CoA reductase inhibitors to lower cholesterol, whereas (R)-CHBE is an important intermediate for L-carnitine, which acts as an antioxidant.3,4 Both enantiomers of 2-chloro-1-(2′,4′-dichlorophenyl)ethanol could be used in the synthesis of antifungal agents, such as miconazole, econazole, and sertaconazole, with different antifungal profiles and activities.5 Hence, the enantioselective preparation of both α-halohydrins enantiomers is of equal importance.The manufacturing industry is searching for efficient, green, energy-saving and environmentally benign procedures.6 The discovery and application of biocatalysts for chemical synthesis, especially for value-added products, promise new synthetic methods due to their specialized and complicated spatial, electronic and polar structures.7 The properties of enzymes mean that they are highly diverse and enantioselective, easy to use, environmentally benign and have a high atom economy.8 Dramatic improvement has been achieved employing enzymes, accompanying the third wave of biocatalysis.9
Because of the demanding diversity in biomanufacturing, there is a constant need for new biocatalysts with altered performance, such as high catalytic efficiency, wide substrate scope, high regio- and enantioselectivity, thermo- and pH-stability and high tolerance to substrates, products, and organic solvents.10 For specific reactions, classical enrichment cultivation is an effective strategy for identifying microorganisms harboring new active enzymes. Based on this, various new and efficient tools have been established to shorten the period for discovering enzymes with desired properties, including metagenomics, shotgun insertion, mutation, genome database mining, and isolation and purification approaches for the original microorganisms.11 Recently, genome database mining, which allows the screening of enzymes with similar sequences through target-reaction-oriented screening and selection, has produced various breakthroughs. Recombinant DNA technology has enabled the rapid increase in accessible genome data (currently at a rate of about 200 genomes per month), providing more and more sequences with unknown functions and promoting the quick identification of naturally-evolved enzyme libraries by genome mining.12 Various strategies have been published for the prediction and rational selection of genes from the genome database.13 However, the focus has not been on strategies to identify robust enantioselective enzymes.
To work as an alternative to chemical synthesis on an industrial scale, one biocatalyst should possess certain properties, such as tolerance to a minimum of 100 g L−1 substrate, >99% enantioselectivity, high operational stability, no byproducts and cofactor addition of <0.1 g L−1.14 Our group has been involved in the discovery and engineering of robust enzymes with promising potential uses in organic synthesis of chiral building blocks with pharmaceutical relevance, as shown in Table 1. An NADH-dependent carbonyl reductase, ScCR, has been identified from Streptomyces coelicolor with a substrate-coupled cofactor regeneration. Its specific production rate was 36.8 gproduct/gdry-cell-weight in the asymmetric preparation of the key chiral precursor for atorvastatin (Lipitor).15 For the synthesis of chiral o-chloromandelic acids, key building blocks for clopidogrel (Plavix), two biocatalysts, an NADPH-dependent ketoreductase, CgKR1, and an novel nitrilase, LaN, have been discovered in Candida glabrata and Labrenzia aggregata, respectively.16,23 Their space-time yield reached >100 g L−1 day−1 for the asymmetric reduction of methyl o-chlorobenzoylformate (CBFM) and enantioselective resolution of o-chloromandelonitrile (CMN), respectively. However, based on our previous results in genome mining, as shown in Table 1, one biocatalyst with desired characteristics was obtained from 8–30 candidates (rejection rate of >87.5%). Nonetheless, most of the genes were not investigated and little is known about their properties because a single screen was run and the selection criteria were generally limited to activity and enantioselectivity towards the target substrate. In our previous work, only one substrate-tolerant carbonyl reductase KtCR was screened out of 30 potential reductases for preparing chiral halohydrins from α-chloro-1-acetophenone (CAPE, 4).18 We expected that the other reductases might display similar or even higher catalytic performance. To rescreen this library for robust reductases, three further rounds of screening were proposed, covering activity and enantioselectivity for substrates with altered substituents as primary screening, operational stability against substrate/product and thermostability. The application potential in the enantioselective preparation of halohydrins was also investigated.
| Enzyme | ECa | Candidateb | Characteristics: substrate, loading [g L−1], ee [%], STY [g L−1 day−1]c | Ref. | |
|---|---|---|---|---|---|
| a EC classification, 1 as oxidoreductase, 3 as hydrolase.b Numbers of candidates.c STY: space-time yield. Substrates: COBE (ethyl 4-chloro-3-oxo-butanate), CBFM (methyl o-chlorobenzoylformate), OPBE (ethyl 3-oxo-phenylbutanate), CAPE (α-chloro-1-acetophenone) QNCO (3-quinuclidinone), CNB (4-cyanonitrobenzene), APA (α-acetoxyphenyl acetate), MDPEA (1-(3′,4′-methylenedioxyphenyl) ethyl ester), CMN (o-chloromandelonitrile). | |||||
| 1 | ScCR | 1 | 10 | COBE, 600, >99, 304 | 15 |
| 2 | CgKR1 | 1 | 8 | CBFM, 300, >98.7, 261 | 16 |
| 3 | CgKR2 | 1 | 13 | OPBE, 206, >99, 700 | 17 |
| 4 | KtCR | 1 | 30 | CAPE, 154, >99, 283 | 18 |
| 5 | ArQR | 1 | 17 | QNCO, 242, >99, 916 | 19 |
| 6 | EsLeuDH | 1 | 15 | TMP, 78.1, >99, 275 | 20 |
| 7 | BaNTR1 | 1 | 24 | CNB, 14.8, >99 sel., 291 | 21 |
| 8 | rPPE01 | 3 | 17 | APA, 64.8, 99, 97.2 | 22 |
| 9 | LaN | 3 | 13 | CMN, 50.2, 96.5, 143 | 23 |
| 10 | BaE | 3 | 22 | MDPEA, 40, 97, 38.6 | 24 |
Results and discussion
Rescreening result with COBE substrate
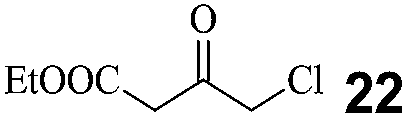
A carbonyl reductase toolbox was developed for robust haloketone reductases. In our primary screening using 4 as the target substrate, only three carbonyl reductases (KtCR, PgCR and ClCR) were returned with higher activity (>0.5 U mg−1) and enantioselectivity (>99%).18 Among them, the highest activity was determined with ClCR (1.5 U mg−1). Further comparison at increased substrate loading revealed the instability of PgCR and ClCR, and only KtCR was selected (Table 2). Most of the candidates were eliminated due to their low activity. However, changing the screening substrate to ethyl 4-chloro-3-oxobutanate (COBE, 22), an α-halogenated β-ketoester, resulted in a different profile (Fig. 1). Twelve reductases were identified with specific activity of more than 0.5 U mg−1, which was higher than that with 4. These exciting results encouraged us to analyze the enantioselectivity in the asymmetric reduction of 22 and 4. DhCR and CgCR, from Debaryomyces hansenii (Uniprot accession no. Q6BQ25) and Candida glabrata (Q6FR42), respectively, displayed stable performance, even at 200 mM of 4 (Table 2). Particularly for DhCR, the time and catalyst requirement was much less than for KtCR, although the apparent specific activity of DhCR was only 38% that of KtCR, indicating that low apparent activity has little connection with the biotransformation efficiency. CgCR was the only reductase with Prelog preference in the asymmetric reduction of prochiral ketones. Reductases with relatively higher activity were more liable to have unstable enantioselectivity. Harsh conditions, such as high reagent concentration (substrate or product) and high temperature, might affect the structural conformation of the enzyme and decrease their activity and enantioselectivity. Considering the activity and ee for two screening substrates, DhCR and CgCR were regarded as interesting reductases, in addition to KtCR. Hence, for the primary screening for activity and operational stability in genome data mining, using at least two substrates with similar structures was better for understanding the enzyme performance comprehensively. However, other characteristics, such as the substrate profiles toward different kinds of substrates and enzyme performance (thermostability), were also considered.| Enzyme | Substrate [mM] | Catalyst [kU L−1] | Time [h] | Conv. [%] | ee [%], R/S |
|---|---|---|---|---|---|
| ClCR | 10 | 1 | 12 | >99 | >99/S |
| 100 | 10 | 12 | >99 | 95.6/S | |
| 200 | 40 | 24 | 80.8 | 80.0/S | |
| KtCR | 10 | 1 | 12 | >99 | >99/S |
| 100 | 10 | 12 | >99 | >99/S | |
| 200 | 20 | 12 | >99 | >99/S | |
| PgCR | 10 | 1 | 12 | >99 | >99/S |
| 100 | 10 | 12 | >99 | 92.4/S | |
| 200 | 40 | 24 | 76.6 | 60.5/S | |
| DhCR | 10 | 1 | 6 | >99 | >99/S |
| 100 | 10 | 6 | >99 | >99/S | |
| 200 | 10 | 6 | >99 | >99/S | |
| CgCR | 10 | 1 | 12 | >99 | 98.7/R |
| 100 | 10 | 12 | >99 | 98.6/R | |
| 200 | 20 | 8 | >99 | 98.7/R |
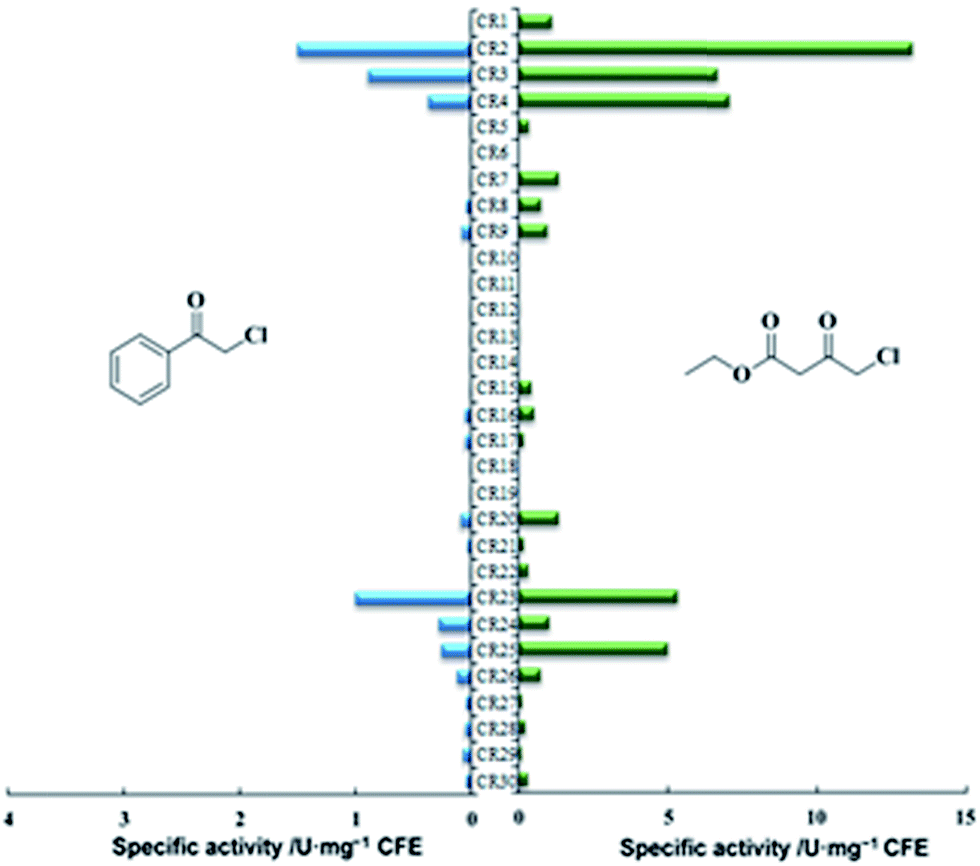 | ||
| Fig. 1 Screening results for the 30 carbonyl reductases using two haloketones (4 and 22) with different substituents. Blue columns: activity towards CAPE, green columns: activity towards COBE. | ||
Enzymatic characteristics of three carbonyl reductases
Three recombinant reductases with an N-terminal His-tag were purified to electrophoretic homogeneity by nickel affinity chromatography. The specific activities toward 22 of purified DhCR, KtCR and CgCR were 13, 11 and 8.0 U mg−1, respectively. Protein separation of the purified enzyme by SDS-PAGE resulted in a single band for each enzyme, corresponding to a molecular weight of 34, 35 and 40 kDa, for DhCR, KtCR and CgCR, respectively (Fig. S1†), in agreement with their theoretical values. Gel exclusion chromatography with a TSK G2000 SWx1 column showed a single peak for DhCR, KtCR and CgCR with an elution volume corresponding to an apparent molecular mass of 67.2 69.6 and 39.8 kDa, respectively, which indicated that they were homodimeric and monomeric enzymes.The effect of pH and temperature on the activity of three carbonyl reductases was investigated (Fig. S2†). All displayed their highest activity at around pH 6.5. The optimum temperatures of DhCR, CgCR and KtCR were 55, 60, and 45 °C, respectively, according to the temperature profiles. The activity of CgCR decreased rapidly above 60 °C due to thermal inactivation. For DhCR and CgCR, the relative activities at 30 °C were 70.8% and 27.0% of the activity at the optimum temperature, respectively. Thermal stabilities were investigated at different temperatures. The half-lives of KtCR, DhCR and CgCR at 30, 40 and 50 °C were 18, 462 and 169 h, 11.8, 111 and 80.6 h, and 0.16, 2.1 and 1.3 h, respectively, as shown in Table 3. DhCR and CgCR were stable at 30 and 40 °C, but unstable at higher temperatures. The deactivation energies (Ea) of DhCR, CgCR and KtCR were 218 ± 6, 198 ± 8, and 190 ± 8 kJ mol−1, respectively. These results indicated that DhCR and CgCR were much more stable at room temperature than at elevated temperatures.26
| Characteristic | KtCR | DhCR | CgCR | |
|---|---|---|---|---|
| Molecular weight [kDa] | 69.8 | 67.2 | 39.8 | |
| Numbers of subunit | 2 | 2 | 1 | |
| Optimal pH | 6.5 | 6.5 | 6.5 | |
| Optimal temperature [°C] | 45 | 55 | 60 | |
| Thermostability [h] | 30 °C | 18.0 | 462 | 169 |
| 40 °C | 11.8 | 111 | 80.6 | |
| 50 °C | 0.163 | 2.09 | 1.26 | |
| Ea [kJ mol−1] | 190 ± 5 | 218 ± 6 | 198 ± 3 | |
 |
KM [mM] | 2.3 ± 0.2 | 2.1 ± 0.1 | 2.2 ± 0.1 |
| kcat [s−1] | 7.3 ± 0.3 | 4.0 ± 0.2 | 1.6 ± 0.1 | |
 |
KM [mM] | 3.2 ± 0.1 | 1.3 ± 0.1 | 3.7 ± 0.2 |
| kcat [s−1] | 13.2 ± 0.2 | 16.6 ± 0.3 | 27.9 ± 0.4 | |
The kinetic constants of the purified reductases for CAPE (4) and COBE (22) were calculated from the Lineweaver–Burk double-reciprocal plot, as shown in Table 3. The kcat for 22 was 16.6 s−1 for DhCR and 27.9 s−1 for CgCR. Relative low KM values for NADPH (<50 μM) for DhCR and CgCR guaranteed high efficiency, even when no external cofactors were added (Table S3†). Although the kcat values for 4 of DhCR and CgCR were lower than that of KtCR, they were much more efficient in the asymmetric reduction of COBE and stable to high temperatures. Their substrate specificity and catalytic performance in the preparation of chiral halohydrins were investigated further.
Substrate profiles of stereocomplementary DhCR and CgCR
No activity with NADH but full activity with NADPH was detected using purified DhCR and CgCR (data not shown), indicating both were NADPH-dependent carbonyl reductases. Twenty-five prochiral ketones (1 to 25) with various substituents, covering aromatic and aliphatic ketones and β-ketoesters, were selected to characterize the substrate spectra of DhCR and CgCR.As shown in Fig. 2, different substrate profiles were observed with DhCR and CgCR. Among the tested substrates, no clear preference was discovered for DhCR, whereas CgCR preferred β-ketoesters to aromatic and aliphatic ketones. In addition to the increase of side-chain length of aromatic ketones, from acetophenone to 1-butyrophenone (1–3), the specific activity of DhCR gradually decreased. CgCR displayed higher catalytic activity for 1-butyrophenone than for 1-propiophenone. Due to the electron-imbalance, α-substituted acetophenone derivatives (4–6, 9–12) were more easily reduced by reductases. Although CN was a strong electron-withdrawing group, benzoylacetonitrile (6) was difficult to reduce because of the steric hindrance of the CN group. 2′-Chloroacetophenone (7) was generally a poor substrate for carbonyl reductases. However, CgCR displayed higher activity toward 7. The specific activity ratio of 4′-chloroacetophenone (8) to 7 for CgCR was 3.2, much lower than 88.3 for DhCR, indicating that CgCR could accept much larger aromatic ketones with ortho substituents on the phenyl ring. Compared with aryl ketones, heteroaryl ketones were more difficult to reduce. Aliphatic ketones were better substrates for DhCR than for CgCR, suggesting that DhCR could be used to prepare chiral aliphatic secondary alcohols. CgCR showed higher activity to β-ketoesters than DhCR, especially to ethyl 4,4,4-trifluro-3-oxo-butonate (23). The highest activity of DhCR was found towards 2,3′,4′-trichloroacetophenone (11, 33 U mg−1). Ethyl 2-chloro-3-oxo-butonate (25, 19 U mg−1) was the best of the tested substrates for CgCR. Interestingly, DhCR showed the opposite enantioselectivity to CgCR, although low enantioselectivity was observed with CgCR for several substrates (20–98% ee, Table S4†). In the asymmetric reduction of prochiral ketones, DhCR obeyed the anti-Prelog rule whereas CgCR complied with Prelog priority. The preparation of both enantiomers of chiral α-halohydrins, which are usually of equal importance, could be achieved with these two stereocomplementary reductases.
 | ||
| Fig. 2 Substrate profiles of DhCR (○) and CgCR (Δ). Specific activities are shown in logarithmic form in the spider web diagram. The activities equal to or lower than 0.01 U mg−1 purified protein are shown as 0.01 U mg−1. Values are listed in Table S4 in ESI.† | ||
In the regions conserved between DhCR and CgCR proteins, which are members of the SDR and AKR families, respectively, typical SDR and AKR sequence motifs, such as cofactor binding, catalytic, substrate binding and structure stabilizing residues, were found, as illustrated in Table S1 and Fig. S4 and S5.†27 This also indicated that DhCR and CgCR belonged to the SDR and AKR families, respectively. Further consensus analysis with homologous proteins showed that they were members of the SDR51C and AKR1B10 subfamilies, based on online database nomenclature searches (http://www.sdr-enzymes.org/, http://www.med.upenn.edu/akr/).28
A characteristic glycine-rich Rossmann-fold scaffold was found in the N-terminal of DhCR for binding the NADP+ dinucleotide.29 The Rossmann-fold sequence in DhCR was TGSSGGIGWA, sharing the motif of the classical or extended subfamily. The length of the extended subfamily is 30 residues longer than the 250 amino acid residues of the classical SDR, and DhCR also had the conserved adenine ring binding and active site motifs of the extended subfamily (Table S2†), DhCR was a member of the extended group.
No Rossmann-fold scaffold motif was found in CgCR. However, as shown in Table S1,† typical motifs for AKR were found in CgCR, which indicated that CgCR was a potential member of AKR superfamily.30 Among the consensus sequences, the catalytic tetrad, Asp-Tyr-Lys-His (50-55-80-111), appeared in the N-terminal of CgCR. Thr26, Asp50, Asn167, Gln190, Ser263 and Arg268 played important roles in cofactor binding. Other residues, such as Gly23, Gly25, Gly45, Asp106, Pro113, Gly165 and Pro187, might play a structural role in forming the barrel core, because they were found in the β-strand, α-helix and short loop regions of the barrel.
Optimization of the asymmetric reduction of COBE to chiral CHBEs
DhCR and CgCR were coexpressed with glucose dehydrogenases separately to provide cofactor regeneration systems. The catalytic performance in the asymmetric reduction of COBE (22) into optically active CHBE with DhCR and CgCR was systematically studied as shown in Table S6.† DhCR could asymmetrically reduce substrate 22 into (S)-CHBE, an important synthon for statin side chains. In contrast, (R)-CHBE was produced with CgCR as an intermediate for L-carnitine 22, and was not stable in the aqueous phase, especially under alkaline conditions, and may be toxic to biocatalysts.31 Biphasic systems are usually used to minimize the loss of the substrate and product. In the toluene/aqueous system, the partition coefficients of 22 and CHBE were 21.3 and 2.8, respectively, and DhCR, CgCR and BmGDH retained their high activity; therefore, toluene was selected as the organic phase.25 The intracellular amount of NADP+ was also quantified to calculate total turnover number (TTN) of these two complementary reductases. There was 1.86 ± 0.13 μmol g−1 NADP+ in DCW of E. coli BL21 (Table S5†), which was slightly higher than the reported amount of 0.39–0.79 μmol g−1 NADP+ in DCW.32 Less than 0.1 μmol g−1 of NADPH was detected, whereas 0.44 ± 0.02 μmol g−1 NAD+ in DCW was calculated in vivo for E. coli BL21. There was less of a difference between E. coli BL21 harbouring DhCR and CgCR coding genes.High substrate loading is desirable for practical applications, usually at more than 100 g L−1. Hence, optimization was carried out to increase the substrate concentration and TTN of reductases in the toluene/aqueous biphasic system. Within 6 h, 0.2 M of 22 was asymmetrically reduced with >99% conversion by using 10 g L−1 DhCR and 5 g L−1 CgCR. In addition to the increase of the substrate/catalyst ratio, as much as 330 g L−1 (660 g L−1 in the toluene phase) of 22 could be fully reduced with no addition of an external cofactor (Table S6†). The preparation of (S)- and (R)-CHBE was scaled up to 1 L, and 330 g of 22 were added to the 0.5 L toluene phase and fully reduced within 24 h (Fig. 3). After purification, the molar isolation yield of (S)- and (R)-CHBE was calculated to be 92.5% and 93.0% respectively, eep was >99%. The substrate/catalyst (S/C) ratio was 33 for CgCR and 16.5 for DhCR.
 | ||
| Fig. 3 Asymmetric reduction of 330 g COBE to both enantiomers of chiral CHBE with DhCR and CgCR in a 1 L reaction mixture (toluene/aqueous biphasic system). | ||
As shown in Fig. 4 and Table S7,† much research has focused on the preparation of (S)-CHBE, because of its widespread use in the preparation of statin side-chains in the pharmaceutical industry. All the reductases that produced (S)-isomers belonged to the SDR family. Except for CmMR from Candida magnolia, most reductases displayed excellent enantioselectivity. The highest substrate loading and TTN of the cofactor were achieved with carbonyl reductase ScCR and CmS1 from Streptomyces coelicolor and C. magnolia.15,31,33 However, the requirement for an external cofactor is a disadvantage for its wide use in organic synthesis.
 | ||
| Fig. 4 Comparison of the catalytic performance of selected ketoreductases in the asymmetric reduction of COBE to chiral CHBE. Black bar (■): total turnover number, red bar (■): space-time yield, blank circle (○): ee. | ||
With no exogenous NADP+, as much as 330 g L−1 of the substrate could be asymmetrically reduced within 24 h using recombinant DhCR, which provided a much more efficient and environmentally friendly reductase for preparing chiral (S)-CHBE. The TTN and space-time yield (STY) of DhCR was 53800 and 305 g L−1 day−1, respectively, calculated based on intracellular NADP+/NADPH. Compared with (S)-isomers, fewer reductases have been reported for producing (R)-CHBE. A carbonyl reductase from Sporobolomyces salmonicolor, SsCR, could catalyze the full reduction of 300 g L−1 of 22, with only 91.7% (R) ee.34 Of the reductases that produce (R)-isomers, the gene YueD from Bacillus subtilis was reported to show the highest ee value at a substrate loading of 214 g L−1 (fed-batch). However, it required 1 mM NADP+,35 which would increase its cost and hinder its application. With no external cofactors, 330 g L−1 of 22 was asymmetrically reduced to optically pure (R)-CHBE, and the S/C, TTN and STY of CgCR were 33108000 and 614 g L−1 day−1, respectively.
Enzymatic preparation of both halohydrin enantiomers
High substrate specificity is an important characteristic of biocatalysts. Different substituents at different positions may imbue substrates with distinct electronic, hydrophobic, polar, and spatial properties. Unlike classical chemical catalysts, there are marked differences between biocatalysts for diverse substrates. The catalytic activity and enantioselectivity may vary according to changes in substrates. An ideal reductase should display high activity, enantioselectivity, and substrate tolerance, and, most importantly, a broad substrate scope. Although it is impossible to find one enzyme for all substrates, as an alternative tool in the organic synthesis of chiral compounds, a series of substrates containing similar functional groups should be catalyzed. Several α-substituted prochiral ketones with high activity and enantioselectivity in the substrate spectra analysis of DhCR and CgCR were chosen to test their applicability.All the tested substrates were reduced at 100 g L−1 substrate loading to optically pure α-halohydrins as illustrated in Table 4. All the α-halohydrins are important chiral building blocks with pharmaceutical relevance. For example, (S)-α-chloro-1-acetophenol could be used for sotalol,36a (S)-2,2,2-trifluroacetophenol for liquid crystals and anticonvulsant pharmaceuticals,36b,c (S)-2,4′-dichloroacetophenol for adrenergic receptor agonists,36d (S)-2,3′,4′-trichloroacetophenol for sertraline,36e (R)-2,2′,4′-trichloroacetophenol for econazole,5 and ethyl (R)-4,4,4-trifluro-3-hydroxybutyrate for befloxatone.36f The efficient preparation of optically pure α-halohydrins shows that DhCR and CgCR are promising catalytic enzymes.
| Substrate | DhCR | CgCR | Chiral blocks in pharmaceutically relevant products | ||||||||
|---|---|---|---|---|---|---|---|---|---|---|---|
| Concn. [g L−1] | Catal.a [g L−1] | Time [h] | Yield [%] | ee [%], R/S | Concn. [g L−1] | Catal.b [g L−1] | Time [h] | Yield [%] | ee [%], R/S | ||
| a Catalyst, dry cells of recombinant E. coli BL21/pET28-bmgdh-dhcr.b Catalyst, dry cells of recombinant E. coli BL21/pET28-bmgdh-cgcr.c Reaction was not performed due to the low ee value (54%).d 330 g L−1 in the reaction mixture and 660 g L−1 in organic phase. | |||||||||||
 |
100 | 30 | 24 | 88 | >99/S | 30.8 | 20 | 24 | 85 | 98/R |  |
 |
174 | 30 | 24 | 90 | >99/S | —c | — | — | — | — |  |
 |
189 | 30 | 24 | 89 | >99/S | 94.5 | 20 | 24 | 90 | >99/R |  |
 |
44.4 | 30 | 12 | 89 | >99/S | 100 | 20 | 12 | 88 | >99/R |  |
 |
100 | 30 | 12 | 90 | >99/S | 100 | 20 | 24 | 87 | >99/R |  |
 |
330 (660)d | 20 | 24 | 92 | >99/S | 330 (660)d | 10 | 12 | 93 | >99/R |  |
 |
184 | 30 | 24 | 89 | >99/S | 184 | 10 | 24 | 92 | >99/R |  |
Conclusions
In summary, two robust haloketone reductases (DhCR and CgCR) were identified from the recently developed carbonyl reductase toolbox through rescreening and characterization. Activity and enantioselectivity are usually the key criteria for chiral biocatalysis. In previous work, the carbonyl reductase KtCR was discovered with high activity and enantioselectivity. However, 29 other candidates were identified by reassessing the enzymatic properties. Hence, a three-round screening strategy was proposed to recheck the missed reductases. Ethyl 4-chloro-3-oxobutanate, a different haloketone from α-chloro-1-acetophenone, was used to retest all 30 reductases. Twelve were returned with activity of more than 0.5 U mg−1. Five with high enantioselectivity were selected to go through the substrate tolerance assay. Only KtCR, DhCR and CgCR were stable enough, and their enzymatic properties were compared. After rescreening their thermostability, DhCR and CgCR, which had opposite enantioselectivity for the asymmetric reduction of prochiral ketones, were identified. At a 1 L scale, both reduced 330 g of 22 into chiral CHBEs, with 92.5% and 93.0% yields, respectively, and >99% ee. The S/C ratios were 33 for CgCR and 16.5 for DhCR. Seven chiral halohydrins with pharmaceutical relevance were asymmetrically prepared. Our results provide key evidence for the stereocomplementary enzymes DhCR and CgCR as potential robust reductases in organic synthesis.Experimental
Materials
Prochiral ketones were all from commercial sources (TCI and Aladdin Inc.). Strains used as genome donors were purchased from CGMCC. The pET28a vector was obtained from Novagen (Madison, WI, USA). Competent cells of E. coli strains, Dh5α and BL21(DE3), were purchased from Tiangen (Shanghai, China).General remarks for gene cloning, expression and purification of proteins
Gene cloning and construction of recombinant plasmids (pET28a-CRs) was reported in our previous work.18 The E. coli BL21(DE3) cells harbouring recombinant pET28a-CRs were cultivated at 37 °C in LB medium containing 50 μg mL−1 kanamycin. When the OD600 of the culture reached 0.5–0.6, IPTG was added to a final concentration of 0.3 mM, and cultivation was continued at 25 °C for a further 15 h. The recombinant E. coli BL21(DE3) cells were collected and purified as previously described.16,18Co-expression of reductase and GDH
The genes coding for KtCR, DhCR and CgCR with independent promoters were separately cut from pET28-ktcr, pET28-dhcr, pET28-cgcr with BglII and XhoI. The pET28-bmgdh vector was digested with BamHI and XhoI. The fragments of ktcr, dhcr and cgcr with independent promoters were ligated with the linear pET28-bmgdh vector to form pET28-bmgdh-ktcr, pET28-bmgdh-dhcr and pET28-bmgdh-cgcr plasmids.25 The resulting plasmids were transformed into E. coli BL21 and over-expressed as mentioned above.Enzyme activity assay
Reductase activity was detected spectrophotometrically at 30 °C through monitoring the change of NAD(P)H absorbance at 340 nm. The reaction mixture consisted of 2 μmol substrate (4 or 22, unless otherwise stated), 0.1 μmol NADPH or NADH, 50 μmol sodium phosphate buffers (pH 6.5), and an appropriate amount of enzyme in a total volume of 1 mL. One unit of enzyme activity was defined as the amount of enzyme that catalyzed the oxidation of 1 μmol of NADPH per minute under these conditions.Enzyme characterization
The optimum pH of KtCR, DhCR and CgCR was determined in the following buffers (final concentration, 50 mM): sodium citrate (pH 4.0–6.0), sodium phosphate (pH 6.0–8.5), and glycine-NaOH (pH 8.5–10.0) using above mentioned enzyme activity assay protocol. The optimum temperature was determined under the standard conditions at various temperatures (25–80 °C). Thermal stability was determined by incubating the purified enzyme (0.1 mg mL−1) at the desired temperature (30, 40 or 50 °C) followed by periodically measuring the residual activity. The kinetic constant analysis of the purified enzyme was performed as previously described.15Conversion and enantioselectivity analysis
The enantioselectivity was determined by examining the reduction of prochiral ketones using an NADPH regeneration system consisting of purified reductase and externally added glucose dehydrogenase (GDH). The reactions were carried out in a reaction mixture (0.4 mL) comprising 50 mM sodium phosphate buffer (pH 6.5), 10 mM carbonyl substrates, 0.2 U of the purified reductase, 0.4 U of BmGDH, 20 mM glucose and 0.5 mM NADP+ with shaking for 12 h at 30 °C and 900 rpm. Each reaction mixture was extracted twice with ethyl acetate. The conversion and ee value were determined by GC analysis equipped with a CP-Chirasil-DEX CB (Varian, USA; 25 m × 0.25 mm × 0.39 mm) column or HPLC analysis using a Chiralcel OD-H column (Daicel Co., Japan; 4.6 × 250 mm) as described in our previous work.16,18Quantification of intracellular NADP+/NADPH
The intracellular amount of NADP+/NADPH of E. coli BL21 was quantified by using HPLC (Shimadzu 2010, Shimadzu Scientific Instruments, Japan) equipped with Shim-pack VP-ODS C18 column (Shimadzu Scientific Instruments, Japan; 4.6 × 250 mm). Dry cells (0.10 g) or wet cells (0.50 g) of E. coli BL21 harbouring pET28a-bmgdh-dhcr or pET28a-bmgdh-cgcr plasmids was weighed and fully dispersed in 10 mL KPB (pH 7.0, 10 mM). The mixture was disrupted with sonication (400 W, 3 s sonication, pause of 7 s) in an ice/water bath and centrifuged at 12000 rpm for 30 min. After that, the upper aqueous phase was filtered, diluted, and injected into the HPLC, which was performed with 3% aqueous acetonitrile containing 0.025 mM N,N,N-triethylamine as the mobile phase at a flow rate of 1.0 mL min−1, monitored at 254 nm, and at 30 °C. The retention times of NAD(H) and NADP(H) were analysed with standards, which were 4.785, 4.555, 7.371 and 8.639 min for NADP+, NAD+, NADH and NADPH, respectively, as shown in Fig. S3.†
General protocol for bioconversion
General protocol for the asymmetric reduction of 4 and 22 into optically active α-chloro-1-acetophenol (CAPL) and ethyl 4-chloro-3-hydroxybutanate (CHBE) was carried out with the whole cell reaction of E. coli harbouring pET28-bmgdh-ktcr, pET28-bmgdh-dhcr and pET28-bmgdh-cgcr. The reaction mixture consisted of 1.0 mmol sodium phosphate buffer (5–10 mL, pH 6.5), 2.0–20.0 mmol of substrates in toluene (5 mL), glucose (1.5 equiv.) and an appropriate amount of dry cells as listed in Table S6.† The reaction was performed by magnetic agitation at 30 °C, 200 rpm and titrated with 2.0 M Na2CO3 to maintain the pH at 6.5 until termination. The reaction mixture was centrifuged (8000 × g for 5 min) to promote phase separation, and then the aqueous phase was saturated with NaCl and extracted with ethyl acetate three times. The organic phase was combined with extraction, dried over anhydrous Na2SO4 and evaporated under vacuum.A 1 L reaction mixture of 20 g coexpressed BmGDH and DhCR or 10 g coexpressed BmGDH and CgCR dry cells, and glucose (30.0 g; 30.0 g portions were added at intervals) in 500 mL sodium phosphate buffer (0.1 M, pH 6.5) and an equal volume of toluene in a 3 L mechanically stirred tank reactor were pre-incubated at 30 °C for 10 min. The reaction was started by adding 330 g of 22. The pH of the reaction mixture was kept at 6.5 with 2.0 M Na2CO3. After stirring at 120 rpm for 24 h, the mixture was extracted with 500 mL ethyl acetate three times. The organic phase was combined, dried over anhydrous Na2SO4 and evaporated under vacuum.
Asymmetric preparation of six α-halohydrins was conducted using the same protocol as COBE (22). The reaction mixture consisted of 1.0 mmol sodium phosphate buffer (5–10 mL, pH 6.5), 2.0–10.0 mmol of substrates in toluene (5 mL), glucose (1.5 equiv.) and appropriate amounts of dry cells as listed in Table 4. The reaction was performed with magnetic agitation at 30 °C, 200 rpm and titrated with 1.0 M Na2CO3 to keep the pH at 6.5 until completion. Reactions were stopped and extracted as above.
Acknowledgements
This work was financially supported by the National Natural Science Foundation of China (no. 21276082), Ministry of Science and Technology, P. R. China (nos 2011CB710800 and 2011AA02A210), China National Special Fund for State Key Laboratory of Bioreactor Engineering (no. 2060204) and Huo Yingdong Education Foundation.Notes and references
- (a) R. N. Patel, ACS Catal., 2011, 1, 1056–1074 CrossRef CAS; (b) J. H. Tao and J. H. Xu, Curr. Opin. Chem. Biol., 2009, 13, 43–50 CrossRef CAS PubMed; (c) J. Magano and J. R. Dunetz, Org. Process Res. Dev., 2012, 16, 1156–1184 CrossRef CAS.
- (a) Z. L. Wei, Z. Y. Li and G. Q. Lin, Tetrahedron, 1998, 54, 13059–13072 CrossRef CAS; (b) D. M. Zhu, C. Mukherjee and L. Hua, Tetrahedron: Asymmetry, 2005, 16, 3275–3278 CrossRef CAS PubMed; (c) N. Itoh, K. Isotani, M. Nakamura, K. Inoue, Y. Isogai and Y. Makino, Appl. Microbiol. Biotechnol., 2012, 93, 1075–1085 CrossRef CAS PubMed.
- H. Yamamoto, K. Mitsuhashi, N. Kimoto, N. Esaki and Y. Kobayshi, Biosci., Biotechnol., Biochem., 2004, 68, 638–649 CrossRef CAS PubMed.
- M. Kataoka, L. P. Rohani, K. Yamamoto, H. Kawabata, M. Wada, K. Kita, H. Yanase and S. Shimizu, Appl. Microbiol. Biotechnol., 1999, 41, 486–490 CrossRef.
- J. Mangas-Sanchez, E. Busto, V. Gotor-Fernandez, F. Malpartida and V. Gotor, J. Org. Chem., 2011, 76, 2115–2122 CrossRef CAS PubMed.
- (a) A. Schmid, J. S. Dordick, B. Hauer, A. Kiener, M. Wubbolts and B. Witholt, Nature, 2001, 409, 258–268 CrossRef CAS PubMed; (b) P. T. Anastas and M. M. Kirchhoff, Acc. Chem. Res., 2002, 35, 686–694 CrossRef CAS PubMed; (c) J. Clark, R. Sheldon, C. Raston, M. Poliakoff and W. Leitner, Green Chem., 2014, 16, 18–23 RSC.
- (a) H. E. Schoemaker, D. Mink and M. G. Wubbolts, Science, 2003, 299, 1694–1697 CrossRef CAS PubMed; (b) R. E. Morris and X. H. Bu, Nat. Chem., 2010, 2, 353–361 CrossRef CAS PubMed; (c) G. W. Zheng and J. H. Xu, Curr. Opin. Biotechnol., 2011, 22, 784–792 CrossRef CAS PubMed.
- (a) K. Nakamura, R. Yamanaka, T. Matsuda and T. Harada, Tetrahedron: Asymmetry, 2003, 14, 2659–2681 CrossRef CAS; (b) H. Gröger, F. Chamouleau, N. Orologas, C. Rollmann, K. Drauz, W. Hummel, A. Weckbecker and O. May, Angew. Chem., Int. Ed., 2006, 45, 5677–5681 CrossRef PubMed; (c) F. Hollmann, I. W. C. E. Arends and D. Holtmann, Green Chem., 2011, 13, 2285–2313 RSC.
- U. T. Bornscheuer, G. W. Huisman, R. J. Kazlauskas, S. Lutz, J. C. Moore and K. Robins, Nature, 2012, 485, 185–194 CrossRef CAS PubMed.
- C. C. C. R. de Carvalho, Biotechnol. Adv., 2011, 29, 75–83 CrossRef CAS PubMed.
- (a) G. A. Behrens, A. Hummel, S. K. Padhi, S. Schatzle and U. T. Bornscheuer, Adv. Synth. Catal., 2011, 353, 2191–2215 CrossRef CAS; (b) T. Davids, M. Schmidt, D. Bottcher and U. T. Bornscheuer, Curr. Opin. Chem. Biol., 2013, 17, 215–220 CrossRef CAS PubMed.
- I. Pagani, K. Liolios, J. Jansson, I. M. A. Chen, T. Smirnova, B. Nosrat, V. M. Markowitz and N. C. Kyrpides, Nucleic Acids Res., 2012, 40, D571–D579 CrossRef CAS PubMed.
- (a) S. Z. Zhu, C. Y. Gong, D. W. Song, S. H. Gao and G. J. Zheng, Appl. Environ. Microbiol., 2012, 78, 7492–7495 CrossRef CAS PubMed; (b) M. Begley, P. D. Cotter, C. Hill and R. P. Ross, Appl. Environ. Microbiol., 2009, 75, 5451–5460 CrossRef CAS PubMed.
- S. Luetz, L. Giver and J. Lalonde, Biotechnol. Bioeng., 2008, 101, 647–653 CrossRef CAS PubMed.
- (a) L. J. Wang, C. X. Li, Y. Ni, J. Zhang, X. Liu and J. H. Xu, Bioresour. Technol., 2011, 102, 7023–7028 CrossRef CAS PubMed; (b) J. Pan, G. W. Zheng, Q. Ye and J. H. Xu, Org. Process Res. Dev., 2014, 18, 739–743 CrossRef CAS.
- H. M. Ma, L. L. Yang, Y. Ni, J. Zhang, C. X. Li, G. W. Zheng, H. Y. Yang and J. H. Xu, Adv. Synth. Catal., 2012, 354, 1765–1772 CrossRef CAS.
- N. D. Shen, Y. Ni, H. M. Ma, L. J. Wang, C. X. Li, G. W. Zheng, J. Zhang and J. H. Xu, Org. Lett., 2012, 14, 1982–1985 CrossRef CAS PubMed.
- G. C. Xu, H. L. Yu, X. Y. Zhang and J. H. Xu, ACS Catal., 2012, 2, 2566–2571 CrossRef CAS.
- W. X. Zhang, G. C. Xu, L. Huang, J. Pan, H. L. Yu and J. H. Xu, Org. Lett., 2013, 15, 4914–4919 Search PubMed.
- J. Li, J. Pan, J. Zhang and J. H. Xu, J. Mol. Catal. B: Enzym., 2014, 105, 11–17 CrossRef CAS PubMed.
- H. H. Nguyen-Tran, G. W. Zheng, X. H. Qian and J. H. Xu, Chem. Commun., 2014, 50, 2861–2864 RSC.
- B. D. Ma, H. L. Yu, J. Pan, J. Y. Liu, X. Ju and J. H. Xu, Bioresour. Technol., 2013, 133, 354–360 CrossRef CAS PubMed.
- C. S. Zhang, Z. J. Zhang, C. X. Li, H. L. Yu, G. W. Zheng and J. H. Xu, Appl. Microbiol. Biotechnol., 2012, 95, 91–99 CrossRef CAS PubMed.
- J. Y. Liu, G. W. Zheng, T. Imanaka and J. H. Xu, Biotechnol. Bioprocess Eng., 2014, 19, 442–448 CrossRef CAS.
- G. C. Xu, H. L. Yu, Z. J. Zhang and J. H. Xu, Org. Lett., 2013, 15, 5408–5411 CrossRef CAS PubMed.
- Y. P. Xu, Y. H. Guan, H. L. Yu, Y. Ni, B. D. Ma and J. H. Xu, J. Mol. Catal. B: Enzym., 2014, 104, 108–114 CrossRef CAS PubMed.
- (a) R. D. Mindnich and T. M. Penning, Hum. Genomics, 2009, 3, 362–370 CAS; (b) B. Persson and Y. Kallberg, Chem.-Biol. Interact., 2013, 202, 111–115 CrossRef CAS PubMed.
- (a) D. Hyndman, D. R. Bauman, V. V. Heredia and M. Penning, Chem.-Biol. Interact., 2003, 143–144, 621–631 CrossRef CAS; (b) B. Persson, Y. Kallberg, J. E. Bray, E. Bruford, S. L. Dellaporta, A. D. Favia, R. G. Duarte, H. Jörnvall, K. L. Kavanagh, N. Kedishvili, M. Kisiela, E. Maser, R. Mindnich, S. Orchard, T. M. Penning, J. M. Thornton, J. Adamski and U. Oppermann, Chem.-Biol. Interact., 2009, 178, 94–98 CrossRef CAS PubMed.
- C. Filling, K. D. Berndt, J. Benach, S. Knapp, T. Prozorovski, E. Nordling, R. Ladenstein, H. Jörnvall and U. Oppermann, J. Biol. Chem., 2002, 277, 25677–25684 CrossRef CAS PubMed.
- L. Di Costanzo, T. M. Penning and D. W. Christianson, Chem.-Biol. Interact., 2009, 178, 127–133 CrossRef CAS PubMed.
- N. Kizaki, Y. Yasohara, J. Hasegawa, M. Wada, M. Kataoka and S. Shimizu, Appl. Microbiol. Biotechnol., 2001, 55, 590–595 CrossRef CAS.
- (a) Z. J. Li, L. Cai, Q. Wu and G. Q. Chen, Appl. Microbiol. Biotechnol., 2009, 83, 929–947 Search PubMed; (b) H. C. Lee, J. S. Kim, W. Jang and S. Y. Kim, J. Biotechnol., 2010, 149, 24–32 CrossRef CAS PubMed.
- Y. C. He, Z. C. Tao, X. Zhang, Z. X. Yang and J. H. Xu, Bioresour. Technol., 2014, 161, 461–464 CrossRef CAS PubMed.
- M. Kataoka, L. P. Rohani, K. Yamamoto, H. Kawabata, M. Wada, K. Kita, H. Yanase and S. Shimizu, Appl. Microbiol. Biotechnol., 1999, 41, 486–490 CrossRef.
- Y. Ni, C. X. Li, L. J. Wang, J. Zhang and J. H. Xu, Org. Biomol. Chem., 2011, 9, 5463–5468 CAS.
- (a) M. Kapoor, N. Anand, K. Ahmad, S. Koul, S. S. Chimni, S. C. Taneja and G. N. Qazi, Tetrahedron: Asymmetry, 2005, 16, 717–725 CrossRef CAS PubMed; (b) T. Fujisawa, K. Ichikawa and M. Shimizu, Tetrahedron: Asymmetry, 1993, 4, 1237–1240 CrossRef CAS; (c) J. Nie, H. C. Guo, D. Cahard and J. A. Ma, Chem. Rev., 2011, 111, 455–529 CrossRef CAS PubMed; (d) H. Lin, Y. Z. Chen, X. Y. Xu, S. W. Xia and L. X. Wang, J. Mol. Catal. B: Enzym., 2009, 57, 1–5 CrossRef CAS PubMed; (e) P. Krumlinde, K. Bogar and J. E. Bäckvall, Chem.–Eur. J., 2010, 16, 4031–4036 CrossRef CAS PubMed; (f) X. Rabasseda, L. A. Sorbera and J. Castaner, Drugs Future, 1999, 24, 1057–1067 CrossRef CAS.
Footnote |
| † Electronic supplementary information (ESI) available. See DOI: 10.1039/c4ra16779a |
| This journal is © The Royal Society of Chemistry 2015 |