
Two-photon absorption and two-photon circular dichroism of hexahelicene derivatives: a study of the effect of the nature of intramolecular charge transfer†
Carlos Díaza,
Yuly Vesgaa,
Lorenzo Echevarriaab,
Irena G. Starác,
Ivo Starỳc,
Emmanuel Angerd,
Chengshuo Shend,
Mehdi El Sayed Moussad,
Nicolas Vanthuynee,
Jeanne Crassousd,
Antonio Rizzof and
Florencio E. Hernández*ag
aDepartment of Chemistry, University of Central Florida, P. O. Box 162366, Orlando, Florida 32816-2366, USA. E-mail: florencio.hernandez@ucf.edu
bDepartamento de Química, Universidad Simón Bolívar, Caracas 1020A, Venezuela
cInstitute of Organic Chemistry and Biochemistry ASCR, Flemingovo nám. 2, CZ-166 10 Prague 6, Czech Republic
dSciences Chimiques de Rennes UMR 6226 CNRS, Universitè de Rennes 1, 35042 Rennes Cedex, France
eAix Marseille Université, Centrale Marseille, CNRS, iSm2 UMR 7313, 13397, Marseille, France
fCNR – Consiglio Nazionale delle Ricerche, Istituto per i Processi Chimico Fisici (IPCF-CNR), UOS di Pisa, Area della Ricerca, Via G. Moruzzi 1, I-56124 Pisa, Italy
gThe College of Optics and Photonics, CREOL, University of Central Florida, P. O. Box 162366, Orlando, Florida 32816-2366, USA
First published on 5th February 2015
Abstract
Herein we report on the theoretical–experimental analysis of the one- and two-photon absorption and circular dichroism spectra of two intrinsically chiral aromatic molecules – hexahelicene derivatives – with helical chirality and intramolecular charge transfer (ICT). The primary outcomes of our investigation demonstrate that the TPA cross-section and the amplitude of the TPCD signal of this type of helicenes are strongly affected by the strength of the ICT and the nature of the extension of the electronic delocalization, i.e. beyond (EXO-ICT) or within (ENDO-ICT) the helicene core. These results were corroborated through the comparative theoretical analysis of the corresponding contributions of the magnetic dipole transition moment and the electric quadrupole transition moment to the TPA rotatory strength on a series of five similar helicene derivatives with different molecular electron delocalization disposition. Two-photon absorption (TPA) and two-photon circular dichroism (TPCD) spectra were obtained using the double L-scan technique over a broad spectral range (400–900 nm) using 90 fs pulses at a low repetition rate (2–50 Hz) produced by an amplified femtosecond system. The theoretical simulations were performed using modern analytical response theory within the Time-Dependent Density Functional Theory (TD-DFT) approach using B3LYP and CAM-B3LYP, and the aug-cc-pVDZ and 6-311++G(d,p) basis sets.
Introduction
The fundamental activity of chiral molecules in biological processes1 and the many applications optically active molecules have found in asymmetric catalysis,2 pharmacy industry,3 and in the world of photonics and nanotechnology,4,5 have motivated scientists to deepen the understanding of chiral systems during the last twenty years. Efforts have been mainly driven by the development of two complementary techniques, i.e. circular dichroism (CD) and optical rotatory dispersion (ORD).6 The former, when associated to transitions between electronic states, is called electronic circular dichroism (ECD) and is defined as the difference in one-photon absorption (OPA) between right (RCPL) and left (LCPL) circularly polarized light.7,8During the last two decades ECD has been a standard technique for the study of the conformational and physical–chemical properties of optically active molecules.6 However, this method presents important intrinsic limitations for the study of optically active molecules in solution and in inhomogeneous media in the UV region.9 The OPA of standard aqueous buffer solutions and common solvents in the same spectral region can mask the ECD signal of chiral molecules and, the scattering present in inhomogeneous samples can become critical at shorter wavelengths.
With the aim of surmounting the existent barriers in ECD, alternative approaches have been proposed; these include vibrational circular dichroism (VCD),10 Raman optical activity (ROA),11 magnetic circular dichroism (MCD),12 and synchrotron radiation circular dichroism (SRCD).13 Likewise, nonlinear optics has opened a new way in this direction by the application of second harmonic generation (SHG),14,15 sum-frequency generation (SFG),15,16 multiphoton optical rotation,17 nonlinear optical activity,18 and two-photon circular dichroism (TPCD).19 The latter is the nonlinear analogue of ECD, which is defined, in the degenerate case, as ΔδTPCD(λ) = δTPAL(λ) − δTPAR(λ), where δTPAL(λ) and δTPAR(λ) are the two-photon absorption (TPA) cross-sections for LCPL and RPCL, respectively, measured at a specific wavelength λ.20–22 TPA is particularly advantageous over OPA in many applications because the typical excitation wavelength for this nonlinear absorption process is approximately two folds longer (degenerate case) than that necessary for OPA. Therefore, the linear absorption in the two-photon excitation region is negligible and scattering can be minimized.23,24 Furthermore, it presents a high intrinsic spatial resolution and penetration depth due to the confinement of the excitation to the focal volume,24 and it exhibits better background discrimination and reduced photodamage.23 Moreover, since experimental measurements of TPA in solution commonly require a relatively high concentration of the solute (typically between 10−3 and 10−2 M), and because the magnitude of the TPCD signal is just a small percentage (1–3%) of δTPA(λ), measurements of TPCD on one specific enantiomer are practically unaffected by the presence of small amounts of the other one. In addition to all the advantages mentioned above for TPA, TPCD is also a very sensitive spectroscopic technique to small structural distortions and with unique access into the far-UV,25 i.e. a spectral region that is filled with important structural/conformational information but typically obscure to ECD. Therefore, the application of TPCD for the structural and conformational study of chiral molecules in this spectral region is virtually imminent. However, before reaching the full potential of this unique approach, more fundamental theoretical–experimental studies on TPCD are required.
Since the development of the double L-scan technique,26 the study of optically active molecules in the near to far UV region using TPCD became an open possibility for the structural study and conformational understanding of chiral systems. It should be mentioned that the first step towards the experimental measurement of TPCD was made by Richardson and co-workers in 1995 (ref. 27) using fluorescence-detected two-photon circular dichroism (FD-TPCD), an approach that inspired Finazzi and co-workers to develop more recently a FD-TPCD based microscopy technique.28
Seeking for a better understanding of TPCD and its structure–property relationship we have been working on the theoretical–experimental analysis of the TPCD response of several systems.19,25,29–33 Although all these studies have contributed to the expanded knowledge of TPCD in organic molecules, more molecular systems need to be studied to better understand this nonlinear optical property and its potential applications. To help accomplish this goal, herein we present the results from our most recent work which was primarily focused on the first theoretical–experimental analysis of the TPA and TPCD spectra of two interesting aromatic molecules – hexahelicene derivatives – with helical chirality and intramolecular charge transfer (ICT) (see Fig. 1a for their chemical structures). One should remark that helicenes are very popular chiral systems employed in applications involving chirooptical photoswitches,34 enantioselective fluorescence detectors,35 circularly polarized luminescence for back-lighting in liquid crystals displays,36,37 and nonlinear optical (NLO) devices.38,39
 | ||
| Fig. 1 (a) Chemical structures of P-(+)-1-aza[6]helicene (A6), and M-(−)-2-[(4-cyanophenyl)-1-ethynyl]carbo[6]helicene (CN6). (b) Chemical structures of CN6-like helicenes (#CN). C6–CN is the base #CN. Derivatives with increasing EXO-ICT and ENDO-ICT are in the upper and lower row, respectively. | ||
Next, we present evidence showing that the TPA cross-section and the amplitude of the TPCD signal in helicenes are strongly affected by the strength and nature of the ICT. We demonstrate that the extension of the electronic delocalization beyond the helicene core (EXO-ICT) has a stronger effect on the nonlinear optical responses of this family of molecules than the extension of the electronic delocalization within the aromatic cluster (ENDO-ICT). This effect is directly correlated, as shown below in a series of helicene derivatives with different electron delocalization disposition (Fig. 1b), with the contributions of the magnetic dipole transition moment and the electric quadrupole transition moment to the TPCD rotatory strength of the molecules.
Experimental details
P-(+)-1-aza[6]helicene (hereafter A6 – Fig. 1a) was synthesized as previously reported by Stará and co-workers.40 Briefly, A6 (Fig. 1a) was prepared by using the cobalt-catalyzed intramolecular [2 + 2 + 2] cycloaddition of a pyridine-linked triyne followed by dehydrogenation with MnO2.40M-(−) and P-(+)-2-[4-(cyanophenyl)-1-ethynyl]carbo[6]helicene (Fig. 1b) were obtained from racemic 2-ethynyl-[6]carbohelicene and commercially available 4-bromobenzonitrile through a Sonogashira coupling reaction followed by chiral HPLC separation.41 Although both the P and M enantiomers were obtained for this derivative, only the M enantiomer (henceforth CN6) was used for the spectroscopic measurements due to impurities that could not be removed from the solution containing the P enantiomer.
OPA measurements were performed using a single-beam spectrophotometer (Agilent 8453 Diode Array UV-Vis) from 190 to 600 nm in a 0.1 cm quartz cell in 0.2 × 10−4 to 2.0 × 10−4 M solutions in THF. ECD spectra were completed on a J-815 CD spectropolarimeter (Jasco Corp., Tokyo, Japan) following these conditions: concentration 1.0 × 10−5 M in THF; temperature 25 °C; quartz cuvette length 4 mm; wavelength range 190–600 nm; 1 nm step and scan speed 50 nm min−1.
TPA and TPCD measurements were carried out in solutions with concentrations ranging from 3.0 × 10−2 M to 5.3 × 10−2 M, employing the double-L scan technique.26 All samples were dissolved in THF. Two-photon excitation was induced with a computer-controlled femtosecond optical parametric amplifier (OPerA Solo) pumped by an amplified laser system from COHERENT. The whole system is capable of generating 90 fs (FWHM) pulses over a wavelength range spanning from 240 nm to 2.6 μm, and with pulse energies of up to 350 μJ. Experiments were performed at a repetition rate between 2 and 50 Hz to avoid any contribution from cumulative effects. The pulse width was measured using a single-shot autocorrelator from Coherent Inc. and a frequency-resolved optical gaiting (FROG) from Swamp Optics LLC.
Theoretical and computational approach
Although, the theoretical approach employed in this work has been extensively addressed elsewhere22,33,42–45 (see also ESI†), below we briefly describe the fundamental aspects of the theoretical and computational calculations.The molecular structures of A6 and CN6 (see Fig. 1a), as well as those of five CN6-like helicenes (#CN) (see Fig. 1b), i.e. C6–CN, C6–![[triple bond, length as m-dash]](https://www.rsc.org/images/entities/char_e002.gif) –CN, C6–––CN, C7–CN and C8–CN, were optimized using Density Functional Theory (DFT),46 employing the Becke's three-parameter exchange, Lee, Yang and Parr correlation (B3LYP) hybrid functional47–49 in combination with the 6-311++G(d,p) basis set,50,51 employing Gaussian 09.52 Solvent effects (THF) were considered as implemented in the polarizable continuum model (PCM).53 Calculations of excited states of all helicenes, in the linear (OPA and ECD) and nonlinear (TPA and TPCD) instance, were performed employing Time-Dependent DFT (TD-DFT),46 with two different exchange–correlation functionals (XCF), i.e. B3LYP and the Coulomb attenuating method-B3LYP (CAM-B3LYP).54 Because it is well-known that the latter is a more reliable XCF than the former in molecules with ICT,29,44,54 in the manuscript we only present results obtained with CAM-B3LYP – results from theoretical calculations using B3LYP are available in ESI.†
–CN, C6–––CN, C7–CN and C8–CN, were optimized using Density Functional Theory (DFT),46 employing the Becke's three-parameter exchange, Lee, Yang and Parr correlation (B3LYP) hybrid functional47–49 in combination with the 6-311++G(d,p) basis set,50,51 employing Gaussian 09.52 Solvent effects (THF) were considered as implemented in the polarizable continuum model (PCM).53 Calculations of excited states of all helicenes, in the linear (OPA and ECD) and nonlinear (TPA and TPCD) instance, were performed employing Time-Dependent DFT (TD-DFT),46 with two different exchange–correlation functionals (XCF), i.e. B3LYP and the Coulomb attenuating method-B3LYP (CAM-B3LYP).54 Because it is well-known that the latter is a more reliable XCF than the former in molecules with ICT,29,44,54 in the manuscript we only present results obtained with CAM-B3LYP – results from theoretical calculations using B3LYP are available in ESI.†
At this point, we would like to comment on the limitations of an inherently single-excitation based model as TD-DFT when dealing with electronic excited states dominated by double excitations.55,56 It is well known that in some cases, for example for polyenes or polyacenes, the TPA response can be particularly intense for states with this character.56,57 Although, we recognize the challenges present when resorting to the computational protocol employed in this work for the study of linear and, in particular nonlinear spectroscopies, this subject has been the theme of detailed studies within our group, cf. for instance ref. 33. Double excitations, in the definition given usually in ab initio quantum chemistry, are somehow taken care even within standard adiabatic TD-DFT approaches using approximate functionals.56,58 Nevertheless, it is a fact that, when double or higher excitation character of the excited state is dominant, this model is bound to fail. Although alternative approaches to adiabatic TD-DFT, able in principle to deal with double excitations, have been proposed and developed (as the use of frequency dependent or non adibatic kernels),55 the debate and the current development has been mostly concerned with excitation energies and linear response properties, and we are not aware of extensions to analytical nonlinear response. On the other hand the importance of multiple excitations for two-photon spectra has been discussed also within the ab initio quantum chemistry community,57 and besides the well-known multiconfigurational time dependent Hartree–Fock approaches to nonlinear response59 available in DALTON60 now for decades – but not suitable in its current status, for the treatment of molecules of the size of those studied here – the algebraic-diagrammatic construction polarization propagator approach developed within the group of Drew and applied to octatetraene in ref. 59, 61 and 62, appears to be a particularly promising suitable tool to treat non-linear absorption when multiple excitation are of particular importance for sizeable molecules.
Next, we computed the oscillator strengths (f0f) and velocity rotatory strengths (RECD0f) for the first 60 electronic excited states for all the helicene derivatives using TD-DFT at the CAM-B3LYP/6-31G(d)63 level of theory including solvent effects (THF) via PCM, in Gaussian 09.52 The convoluted theoretical UV-vis and ECD spectra were obtained by Lorentzian broadening from f0f and RECD0f of the calculated electronic transitions employing eqn (1) and (2). Note: in order to certify the reliability of the selected basis set, we performed additional TD-DFT calculations over a smaller number of excited states (first 30 electronic excited states) using a larger basis set, i.e. 6-311++G(d,p). No significant differences, compared to the linear spectra computed with 6-31G(d) were obtained.
TPA probabilities  and TPCD rotatory strength (RTPCD0f(ω0f)) for all the helicene derivatives were calculated employing TD-DFT in DALTON 2011,60 using CAM-B3LYP/aug-cc-pVDZ64 for A6, and employing CAM-B3LYP/6-311++G(d,p) for the rest of the helicene derivatives. For TPA and TPCD, the number of electronic excited states selected for A6 (60 & 48) and CN6 (60 & 40), were determined by the experimental spectral range (200–450 nm) covered during the measurements. In #CN derivatives the number of electronic excited states to be considered in the calculations were established by the measured experimental spectral range for CN6 (see captions for Fig. 4 and 5 for the accurate number of electronic excited states employed for each derivative). No solvent effects were considered in the nonlinear calculations due to the high computational costs. The convoluted theoretical TPA and TPCD spectra were obtained by Lorentzian broadening from
and TPCD rotatory strength (RTPCD0f(ω0f)) for all the helicene derivatives were calculated employing TD-DFT in DALTON 2011,60 using CAM-B3LYP/aug-cc-pVDZ64 for A6, and employing CAM-B3LYP/6-311++G(d,p) for the rest of the helicene derivatives. For TPA and TPCD, the number of electronic excited states selected for A6 (60 & 48) and CN6 (60 & 40), were determined by the experimental spectral range (200–450 nm) covered during the measurements. In #CN derivatives the number of electronic excited states to be considered in the calculations were established by the measured experimental spectral range for CN6 (see captions for Fig. 4 and 5 for the accurate number of electronic excited states employed for each derivative). No solvent effects were considered in the nonlinear calculations due to the high computational costs. The convoluted theoretical TPA and TPCD spectra were obtained by Lorentzian broadening from  and RTPCD0f(ω0f) of the calculated electronic transitions employing eqn (3) and (4). This step was completed using Lorentzian lineshape functions g(ω,ω0f,Γ) and g(2ω,ω0f,Γ) for the one- and two-photon cases, respectively, with half width at half maximum (FWHM) Γ, and centered on the computed excitation circular frequency ω0f for a 0 → f transition. All the OPA and ECD spectra were obtained using a linewidth (Γ) of 0.5 eV (FWHM). For the TPA and TPCD spectra a value of Γ = 0.15 eV (FWHM) was used.
and RTPCD0f(ω0f) of the calculated electronic transitions employing eqn (3) and (4). This step was completed using Lorentzian lineshape functions g(ω,ω0f,Γ) and g(2ω,ω0f,Γ) for the one- and two-photon cases, respectively, with half width at half maximum (FWHM) Γ, and centered on the computed excitation circular frequency ω0f for a 0 → f transition. All the OPA and ECD spectra were obtained using a linewidth (Γ) of 0.5 eV (FWHM). For the TPA and TPCD spectra a value of Γ = 0.15 eV (FWHM) was used.
OPA spectra are reported in molar absorptivity (ε),45,65
 | (1) |
ECD spectra were computed from RECD0f, and they are reported as molar absorptivity difference (Δε),45,65,66
 | (2) |
OPA and ECD spectra have units of mol−1 cm−1 l, as long as all the elements in eqn (1) and (2) are introduced in atomic units.
TPA spectra were obtained from65,67
 | (3) |
![[small delta, Greek, macron]](https://www.rsc.org/images/entities/i_char_e0c5.gif) TPA0f(ω0f) is the orientationally averaged two-photon probability for the degenerate case.
TPA0f(ω0f) is the orientationally averaged two-photon probability for the degenerate case.
TPCD spectra were simulated according to,20,42,65
 | (4) |
TPA and TPCD spectra obtained from eqn (3) and (4) are given in Göppert-Mayer units (GM), i.e., 10−50 cm4 s per molecule per photon, as long as all the equation elements are introduced in atomic units.
Results and discussion
In order to start with the analysis, in Fig. 2 we present the experimental OPA and ECD spectra of A6 (Top) and CN6 (Bottom) in THF solution, and their corresponding convoluted CAM-B3LYP stick spectra, calculated in THF using PCM and basis set 6-31G(d) (assignment of theoretical excited states to experimental bands can be found in Tables 1a and b in ESI†). To obtain a better match with the experiment, the theoretical spectra were spectrally shifted by an amount reported in the caption of the figure (this is common practice in theoretical–experimental works).19,29,30,32,33 One should mention that the theoretical curves are consistently blue shifted with respect to the experimental. This result is not surprising since, as it has been previously reported,19,33,45,68 in all calculations most of the depicted states are basically valence states with a significant contribution form π → π*, and because CAM-B3LYP tends to overestimate the excitation energies.33 | ||
| Fig. 2 Experimental (black solid line) and theoretical (colored dotted lines) UV-vis (left column) and ECD (right column) spectra of A6 and CN6. Colored empty symbols display the oscillator strengths for each molecule. OPA for the lowest 60 electronic excited states were computed at the CAM-B3LYP/6-31G(d) level of theory using Gaussian 09 in THF and employing PCM. The theoretical spectra are only shown within the measurable spectral range (200 nm through 450 nm) with spectral shifts: A6 (+20 nm) and CN6 (0 nm). Γ = 0.5 eV (FWHM) was used for all the spectra. Excited states contributing to 20% or more of the total intensity of prominent spectral features observed in the theory and the experiment are highlighted. All the experimental spectra were taken in THF solutions. | ||
The primary observation in Fig. 2 is the noteworthy theoretical reproduction of the experimental OPA and ECD spectra (spectral shape, position of the bands, and the fano-type shape profile) of A6 and CN6 using CAM-B3LYP. The remarkably good performance of this particular XCF is attributed to the existent intramolecular charge transfer (ICT) in both molecules, being ICT unequivocally stronger in CN6. In this particular molecule, the cyano (–CN) group, a well-known moderate electron-withdrawing group, located beyond the helicene core on one extreme of the molecule, causes not only a strong ICT effect but it increases the range of the electronic delocalization beyond the helicene core. This point can be confirmed even further by the analysis presented in Fig. 3, which show the molecular orbitals involved in the transitions to excited state #3 and #2 for A6 and CN6, respectively, i.e. the most important excitations on the red side of the OPA and ECD theoretical spectra calculated with CAM-B3LYP (see Fig. 2 and Tables 1a and b in ESI†). The observed ICT in all three excitations involved in the CN6 transition is indeed extremely important. Similar evidences have already been reported by Rizzo and co-workers in other molecular systems29,33,45,68 and, are in accordance with previous calculations performed in CN6.41
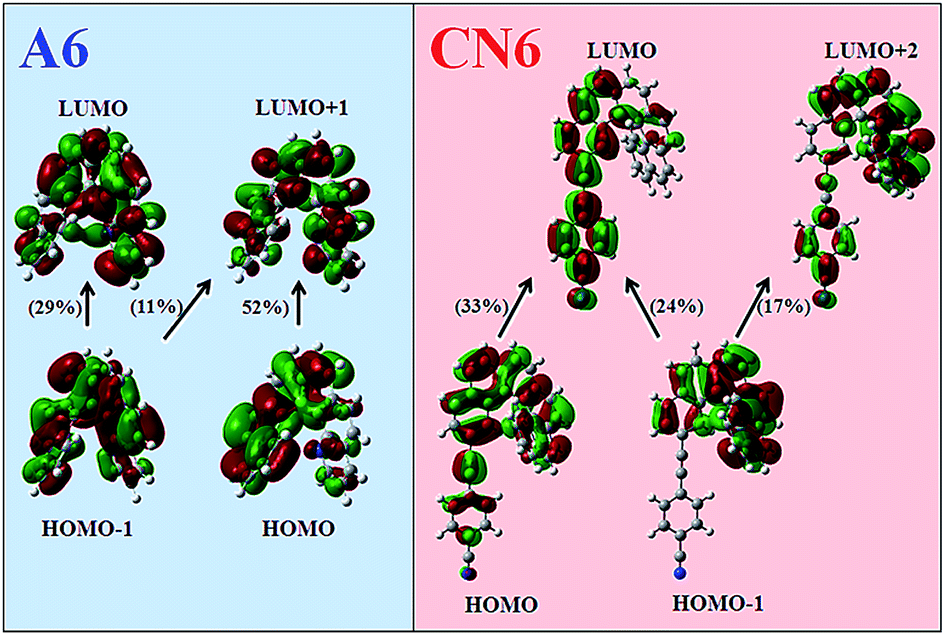 | ||
| Fig. 3 Molecular orbitals (MOs) involved in the 3rd and 2nd electronic excited state of A6 and CN6, respectively. The MOs were obtained from CAM-B3LYP/6-31G* TD-DFT calculations in THF using PCM in Gaussian 09. The percent contribution from single excitations ([HOMO − x] → [LUMO + y]) to the excited state is indicated in parenthesis for each case. | ||
Next, one should highlight the fact that the amplitudes of the OPA and ECD signals in both molecules are virtually identical, though there is an obvious difference in ICT between the two. This observation shall be explained more in detail after completing the theoretical–experimental analysis of the nonlinear spectra of A6 and CN6 presented next.
In Fig. 4 we show the TPA and TPCD spectra (theoretical and experimental), respectively, plotted at half the excitation wavelengths, i.e. in the OPA scale. The spectra of both compounds were taken in THF solution. The theoretical spectra of A6 were obtained with the CAM-B3LYP/aug-cc-pVDZ computed profiles obtained by convoluting the stick spectra (the latter simulates isolated non interacting molecules). To optimize the balance between accuracy and computational cost, calculations in CN6 were performed using the 6-311++G(d,p) basis set, which is a slightly smaller basis set than aug-cc-pVDZ. As for the one-photon spectra a shift has been applied to the computed TPA and TPCD spectra (see figure caption of Fig. 4). Since the spectroscopic region of relevance is different when comparing one- and two-photon absorption profiles, the shifts applied in Fig. 4, chosen to improve the comparison between theory and experiment, are different from those applied in Fig. 2 (assignment of theoretical CAM-B3LYP excited states to experimental bands of A6 and CN6 can be found in Tables 1a and b in ESI†). Also, because strong two-photon absorber states are in most cases weak one-photon absorber, the differences in spectral shift obtained for OPA and TPA are easily explained by considering that OPA and TPA are dominated (see Fig. 2 and 4) by different molecular states.69,70
 | ||
| Fig. 4 Experimental (black scattered squares) and theoretical TPA (left column) and TPCD (right column) spectra of A6 and CN6 calculated in vacuo using Dalton 2011. TPA was computed for the first 60 electronic excited states (colored scattered symbols) for both molecules. TPCD was computed for the first 48 and 40 electronic excited states (colored scattered symbols) for A6 and CN6, respectively. The Lorentzian convolution (colored dotted lines) was obtained using a linewidth Γ = 0.15 eV (FWHM). The theoretical spectra were calculated with CAM-B3LYP/aug-cc-pVDZ for A6, and CAM-B3LYP/6-311++G(d,p) for CN6. The theoretical spectral shifts are: A6 (+22 nm) and CN6 (+26 nm). Excited states contributing to 20% or more of the total intensity of prominent spectral features observed in the theory and the experiment are highlighted. All the experimental spectra were taken in THF solutions. | ||
First, it can be noticed that the theoretical calculations reproduce remarkably the main TPA spectral features of both helicene derivatives. However, the predicted TPA cross-sections are, in most cases, larger than the experimental. It is hard to explain this difference in amplitude considering that all experiments were performed in the femtosecond regime where excited state absorption is negligible.71,72 Nevertheless, the chosen XCF reproduces very well the relative intensities between the main TPA bands, and it yields a suitable bandwidth and separation between the peaks.
Second, the intense peak observed at 215 nm in the TPA experimental spectrum of A6, that lead to a strong increasing band in the blue side of the TPA spectrum of this molecule is not well reproduced by the theory. This outcome could be a consequence of the limited number of electronic excited states included in the theoretical calculation of the TPA spectrum or, even more probable, due to the presence of strong TPA resonance enhancement effect found when the measurements are performed in the close vicinity of linear absorption states (see Fig. 2).73,74 In any case, this region would be better analysed with the damped approach to TPA discussed in ref. 67 and 68.75
Next, moving onto TPCD, in Fig. 4 we display the experimental (in THF solution) and theoretical (isolated molecules) spectra of A6 and CN6, obtained using CAM-B3LYP. One can notice that the theoretical TPCD spectra of A6 and CN6 reproduces notably the shape, sign, and relative intensities of the main three bands in A6 (one positive at 240 nm and two negative at 250 and 290 nm) and the three positive bands in CN6 (280, 300 and 330 nm) – see Tables 1a and b in ESI.†
Finally and perhaps the most interesting outcome of the nonlinear characterization of these two molecules is the obviously larger TPA cross-section (approximately ten times) and TPCD signal (ca. two-fold) of CN6 compared to that of A6 (see Fig. 4). This interesting result can be explained by considering two cooperative effects: (a) the strong ICT character of a well-known moderate electron-withdrawing group (–CN) on one end of CN6, and (b) the extended electron delocalization over the (4-cyanophenyl)-1-ethynyl substituent outside the helical core in this helicene derivative. In A6, however, a relatively mild ICT is effective only within the hexahelicene core due to the presence of the aza (–N![[double bond, length as m-dash]](https://www.rsc.org/images/entities/char_e001.gif) ) group at one end of the molecule. Although, the difference in ICT between A6 and CN6 was discussed earlier in the linear characterization of these two molecules (see Fig. 4), the actual implications were not elucidated.
) group at one end of the molecule. Although, the difference in ICT between A6 and CN6 was discussed earlier in the linear characterization of these two molecules (see Fig. 4), the actual implications were not elucidated.
In order to gain more insight about the two effects mentioned above, we performed calculations of OPA, ECD, TPA and TPCD on five #CN molecules (see Fig. 1b) exhibiting different ICT conjugation length outside the [6]carbohelicene core (EXO-ICT), i.e. C6–CN, C6––CN and C6–––CN, and within the aromatic cluster (ENDO-ICT), i.e. carbohelicene derivatives with six (C6–CN), seven (C7–CN) and eight (C8–CN) aromatic rings extending the helical arrangement.
In Fig. 5 we show the comparative plots of the OPA and ECD, and the TPA and TPCD theoretical spectra of all five #CN. As expected from the linear analysis of A6 and CN6, the OPA and ECD signals are not strongly affected by the nature of ICT, i.e. EXO-ICT vs. ENDO-ICT. In this figure one can only observe a small increase in the amplitude of the ECD signal and a consistent red shift of the position of the fano-type bands of #CN as the conjugation length of the aromatic cluster increases from C6 → C8. However, EXO-ICT seems to have no significant effect on the linear spectra of these molecules. This interesting result suggests the generation of stronger molecular circular currents as the spiral length of the ENDO-ICT increase in helical molecules. These currents can in turn generate molecular magnetic fields that couple with the magnetic dipole transition moment of #CN,32,76,77 thus increasing the ECD response as follow: C6–CN < C7–CN < C8–CN.
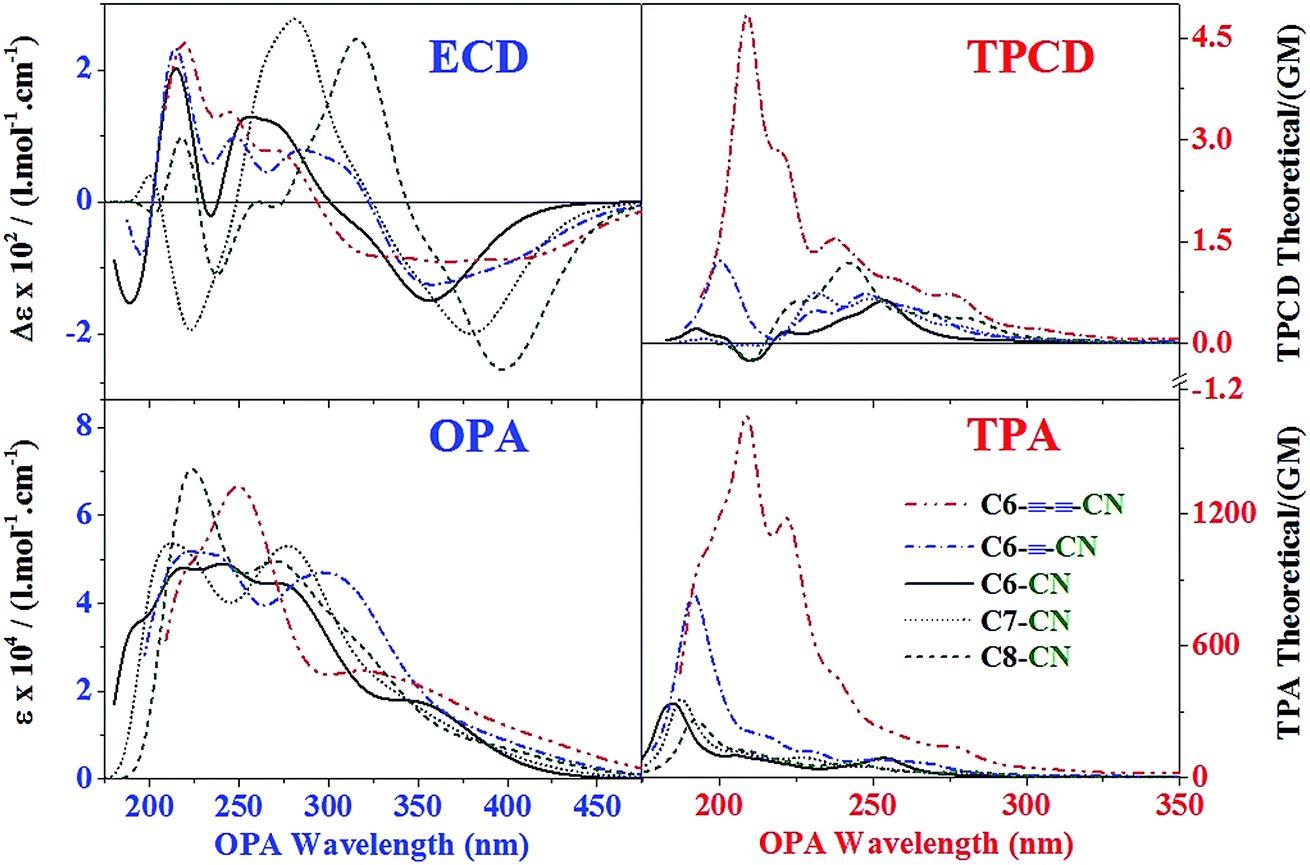 | ||
| Fig. 5 OPA, ECD, TPA and TPCD theoretical spectra of all five #CN. OPA and ECD spectra were obtained from the Lorentzian convolution (Γ = 0.5 eV FWHM) of the first 60 excited states excited states of the optimized structures of all five #CN calculated at the CAM-B3LYP/6-31G(d) level of theory in THF using PCM and employing Gaussian 09. TPA spectra of the optimized structures of all five #CN derivatives were obtained from the Lorentzian convolution (Γ = 0.15 eV FWHM) of the first 60 excited states excited states except for C7–CN (67 states) and C8–CN (73 states). TPCD spectra of the optimized structures of all five #CN derivatives were obtained from the Lorentzian convolution (Γ = 0.15 eV FWHM) of the first 40 excited states except for C7–CN (45 states). TPA and TPCD calculations were performed at the CAM-B3LYP/6-311++G(d,p) level of theory in vacuo using Dalton 2011. | ||
In the nonlinear optical response instance it is obvious that the effect of EXO-ICT is much stronger than ENDO-ICT. In Fig. 5, one can notice that while the maximum TPA cross-sections of C6––CN and C6–––CN are approximately 2.4 and 4.9 times that of C6–CN, respectively, the difference in TPA between C6–CN, C7–CN and C8–CN is nearly negligible – the small difference noticed between C6–CN, C7–CN and C8–CN, on the blue side of the spectra, is mainly attributed to the limitations of our computational resources to calculate the required number of excited states in the larger molecules (C7–CN and C8–CN) to cover the same spectral region obtained for C6–CN. In TPCD one can also remark a stronger EXO-ICT effect, i.e. the integrated area of the TPCD spectra of C6––CN and C6–––CN are approximately 2.5 and 7.9 folds that of C6–CN, respectively. The difference in TPCD between C7–CN and C8–CN, and C6–CN is ca. 1.7 and 2.2, respectively, i.e. less significant than in the previous series of #CN but comparable to the small differences observed in ECD. This results strongly suggest that: (1) the electric dipole transition moment, which directly affect the TPA response of conjugated molecules,78,79 is enhanced by increasing the electronic conjugation length beyond the helicene core, (2) the magnetic dipole transition moment could enhance the TPCD response of helical molecules via its coupling with the induced molecular magnetic field resultant from the generation of molecular circular currents within the helical core,32,76,77 and (3) the contribution of the electric quadrupole transition moment to the TPCD signal of helical molecules with strong EXO-ICT – the extended electronic conjugation beyond the helicene core significantly enhances the nonlinear response of this type of molecular structures. While points (2) and (3) are crucial for TPCD in the series of molecules with greater EXO-ICT (C6–CN, C6––CN and C6–––CN), in the other series of molecules (C6–CN, and C7–CN and C8–CN) point (2) becomes more critical.
With the intention of supporting even further these results, in Fig. 6 we present the integrated TPCD spectra, and the sum of the absolute values of molecular parameters B1 + B3 and B2 that define the contributions of the magnetic dipole transition moment and the electric quadrupole transition moment, respectively, to the TPCD rotatory strength20,22,42 (see ESI† for further detail on molecular parameters Bi). The clear tendencies observed in this plot, viz. (i) ∑|B1 + B3| (Fig. 6b) following the same trend observed in TPCD (Fig. 6a) within the whole series of #CN, and (ii) ∑|B2| (Fig. 6c) matching only the EXO-ICT series (C6–CN, C6––CN and C6–––CN) confirm our initial thoughts.
 | ||
| Fig. 6 Comparative bar graph of (a) integrated TPCD spectra, (b) ∑|B1 + B3| and (c) ∑|B2|, for all five #CN. | ||
Conclusions
In summary, we have demonstrated that the TPA cross-section and the amplitude of the TPCD signal in helicenes are strongly affected not only by the strength of the ICT but by its nature. The molecular orbital analysis of A6 and CN6, and the theoretical analysis of these effects on a series of CN6-like helicenes validated our hypothesis. Through this research we were able to show that while the extension of the electronic delocalization beyond the helicene core (EXO-ICT) enhances the contribution of the electric quadrupole and magnetic transition moments to the TPCD rotatory strength (RTPCD0f(ω0f)), the electronic delocalization within the arrangement of aromatic rings (ENDO-ICT) primarily increases the contribution of the magnetic dipole transition moment to RTPCD0f(ω0f).Acknowledgements
Jeanne Crassous thanks the Ministère de l'Education Nationale, de la Recherche et de la Technologie and the Centre National de la Recherche Scientifique (CNRS). This work has been partially supported by the National Science Foundation through Grant Number NSF/ARRA-0840431 and NSF/DBI-1422826. The extended computing time provided by STOKES ARCC/UCF is gratefully acknowledged.References
- K. Tamura, BioSystems, 2008, 92, 91–98 CrossRef CAS PubMed.
- R. Noyori, Angew. Chem., Int. Ed., 2002, 41, 2008–2022 CrossRef CAS.
- O. McConnell, A. Bach, C. Balibar, N. Byrne, Y. Cai, G. Carter, M. Chlenov, L. Di, K. Fan and I. Goljer, et al., Chirality, 2007, 19, 658–682 CrossRef CAS PubMed.
- J. Zhang, M. T. Albelda, Y. Liu and J. W. Canary, Chirality, 2005, 17, 404–420 CrossRef CAS PubMed.
- L. M. Haupert and G. J. Simpson, Annu. Rev. Phys. Chem., 2009, 60, 345–365 CrossRef CAS PubMed.
- K. Nakanishi, N. Berova and R. W. Woody, Circular Dichroism: Principles and Applications, VCH, New York, 2nd edn, 2000 Search PubMed.
- E. U. Condon, Rev. Mod. Phys., 1937, 9, 432–457 CrossRef CAS.
- D. J. Caldwell and H. Eyring, The Theory of Optical Activity, Wiley-Interscience, New York, 1971 Search PubMed.
- I. Tinoco, Jr, W. Mickols, M. F. Maestre and C. Bustamante, Annu. Rev. Biophys. Biophys. Chem., 1987, 16, 319–349 CrossRef PubMed.
- L. A. Nafie, Vibrational Optical Activity: Principles and Applications, John Wiley & Sons, West Sussex, 2011 Search PubMed.
- L. D. Barron, Molecular Light Scattering and Optical Activity, Cambridge University Press, Cambridge, 2004 Search PubMed.
- W. R. A. Mason, Practical Guide to Magnetic Circular Dichroism Spectroscopy, John Wiley & Sons, New Jersey, 2007 Search PubMed.
- A. J. Miles and B. A. Wallace, Chem. Soc. Rev., 2006, 35, 39–51 RSC.
- A. Rizzo and H. Agren, Phys. Chem. Chem. Phys., 2013, 15, 1198–1207 RSC.
- P. Fischer and F. Hache, Chirality, 2005, 17, 421–437 CrossRef CAS PubMed.
- N. Ji, V. Ostroverkhov, M. Belkin, Y. J. Shiu and Y. R. Shen, J. Am. Chem. Soc., 2006, 128, 8845–8848 CrossRef CAS PubMed.
- R. Cameron and G. C. Tabisz, Mol. Phys., 1997, 90, 159–164 CrossRef CAS.
- M. Ren, E. Plum, J. Xu and N. I. Zheludev, Nat. Commun., 2012, 3, 833 CrossRef PubMed.
- C. Toro, L. De Boni, N. Lin, F. Santoro, A. Rizzo and F. E. Hernandez, Chem.–Eur. J., 2010, 16, 3504–3509 CrossRef CAS PubMed.
- I. Tinoco, J. Chem. Phys., 1975, 62, 1006–1009 CrossRef CAS PubMed.
- E. A. Power, J. Chem. Phys., 1975, 63, 1348–1350 CrossRef CAS PubMed.
- F. E. Hernández and A. Rizzo, Molecules, 2011, 16, 3315–3337 CrossRef PubMed.
- W. Denk, J. Strickler and W. Webb, Science, 1990, 248, 73–76 CAS.
- B. H. Cumpston, S. P. Ananthavel, S. Barlow, D. L. Dyer, J. E. Ehrlich, L. L. Erskine, A. A. Heikal, S. M. Kuebler, I.-Y. S. Lee and D. McCord-Maughon, et al., Nature, 1999, 398, 51–54 CrossRef CAS PubMed.
- Y. Vesga, C. Diaz, M. Higgs and F. E. Hernandez, Chem. Phys. Lett., 2014, 601, 6–12 CrossRef CAS PubMed.
- L. De Boni, C. Toro and F. E. Hernandez, Opt. Lett., 2008, 33, 2958–2960 CrossRef.
- K. E. Gunde and F. S. Richardson, Chem. Phys., 1995, 194, 195–206 CrossRef CAS.
- M. Savoini, X. Wu, M. Celebrano, J. Ziegler, P. Biagioni, S. C. J. Meskers, L. Duò, B. Hecht and M. Finazzi, J. Am. Chem. Soc., 2012, 134, 5832–5835 CrossRef CAS PubMed.
- C. Díaz, L. Echevarria, A. Rizzo and F. E. Hernández, J. Phys. Chem. A, 2014, 118, 940–946 CrossRef PubMed.
- C. Díaz, L. Echevarria and F. E. Hernández, J. Phys. Chem. A, 2013, 117, 8416–8426 CrossRef PubMed.
- C. Diaz, L. Echevarria and F. E. Hernández, Chem. Phys. Lett., 2013, 568–569, 176–183 CrossRef CAS PubMed.
- C. Diaz, N. Lin, C. Toro, R. Passier, A. Rizzo and F. E. Hernández, J. Phys. Chem. Lett., 2012, 3, 1808–1813 CrossRef CAS.
- N. Lin, F. Santoro, X. Zhao, C. Toro, L. De Boni, F. E. Hernández and A. Rizzo, J. Phys. Chem. B, 2011, 115, 811–824 CrossRef CAS PubMed.
- T. J. Wigglesworth, D. Sud, T. B. Norsten, V. S. Lekhi and N. R. Branda, J. Am. Chem. Soc., 2005, 127, 7272–7273 CrossRef CAS PubMed.
- M. T. Reetz and S. Sostmann, Tetrahedron, 2001, 57, 2515–2520 CrossRef CAS.
- J. E. Field, G. Muller, J. P. Riehl and D. Venkataraman, J. Am. Chem. Soc., 2003, 125, 11808–11809 CrossRef CAS PubMed.
- R. Hassey, E. J. Swain, N. I. Hammer, D. Venkataraman and M. D. Barnes, Science, 2006, 314, 1437–1439 CrossRef CAS PubMed.
- T. Verbiest, S. V. Elshocht, M. Kauranen, L. Hellemans, J. Snauwaert, C. Nuckolls, T. J. Katz and A. Persoons, Science, 1998, 282, 913–915 CrossRef CAS.
- T. Verbiest, S. Sioncke, A. Persoons, L. Vyklicky and T. J. Katz, Angew. Chem., Int. Ed., 2002, 41, 3882–3884 CrossRef CAS.
- J. Míšek, F. Teplý, I. G. Stará, M. Tichý, D. Šaman, I. Císařová, P. Vojtíšek and I. Starý, Angew. Chem., Int. Ed., 2008, 47, 3188–3191 CrossRef PubMed.
- M. E. S. Moussa, M. Srebro, E. Anger, N. Vanthuyne, C. Roussel, C. Lescop, J. Autschbach and J. Crassous, Chirality, 2013, 25, 455–465 CrossRef CAS PubMed.
- B. Jansik, A. Rizzo and H. Agren, Chem. Phys. Lett., 2005, 414, 461–467 CrossRef CAS PubMed.
- B. Jansík, A. Rizzo and H. Agren, J. Phys. Chem. B, 2007, 111, 446–460 CrossRef PubMed.
- N. Lin, F. Santoro, X. Zhao, A. Rizzo and V. Barone, J. Phys. Chem. A, 2008, 112, 12401–12411 CrossRef CAS PubMed.
- M. Guillaume, K. Ruud, A. Rizzo, S. Monti, Z. Lin and X. Xu, J. Phys. Chem. B, 2010, 114, 6500–6512 CrossRef CAS PubMed.
- E. Runge and E. K. U. Gross, Phys. Rev. Lett., 1984, 52, 997–1000 CrossRef CAS.
- A. D. Becke, Phys. Rev. A, 1988, 38, 3098–3100 CrossRef CAS.
- A. D. Becke, J. Chem. Phys., 1993, 98, 5648–5652 CrossRef CAS PubMed.
- C. Lee, W. Yang and R. G. Parr, Phys. Rev. B: Condens. Matter Mater. Phys., 1988, 37, 785–789 CrossRef CAS.
- R. Krishnan, J. S. Binkley, R. Seeger and J. A. Pople, J. Chem. Phys., 1980, 72, 650–654 CrossRef CAS PubMed.
- T. Clark, J. Chandrasekhar, G. W. Spitznagel and P. V. R. Schleyer, J. Comput. Chem., 1983, 4, 294–301 CrossRef CAS.
- M. J. Frisch, G. W. Trucks, H. B. Schlegel, G. E. Scuseria, M. A. Robb, J. R. Cheeseman, G. Scalmani, V. Barone, B. Mennucci and G. A. Petersson, et al., Gaussian 09 Revision A.1, Gaussian, Inc., Wallingford CT, 2009 Search PubMed.
- J. Tomasi, B. Mennucci and R. Cammi, Chem. Rev., 2005, 105, 2999–3093 CrossRef CAS PubMed.
- T. Yanai, D. P. Tew and N. C. Handy, Chem. Phys. Lett., 2004, 393, 51–57 CrossRef CAS PubMed.
- M. A. Marques, N. T. Maitra, F. M. Nogueira, E. K. Gross and A. Rubio, Fundamentals of time-dependent density functional theory, Springer, 2012 Search PubMed.
- C. A. Ullrich, Time-dependent density-functional theory: concepts and applications, Oxford University Press, 2011 Search PubMed.
- K. Aryanpour, A. Roberts, A. Sandhu, R. Rathore, A. Shukla and S. Mazumdar, J. Phys. Chem. C, 2014, 118, 3331–3339 CAS.
- S. Hirata and M. Head-Gordon, Chem. Phys. Lett., 1999, 302, 375–382 CrossRef CAS.
- J. Olsen and P. Jo, J. Chem. Phys., 1985, 82, 3235–3264 CrossRef CAS PubMed.
- K. Aidas, C. Angeli, K. L. Bak, V. Bakken, R. Bast, L. Boman, O. Christiansen, R. Cimiraglia, S. Coriani and P. Dahle, et al., WIREs Computational Molecular Science, 2014, 4, 269–284 CrossRef CAS PubMed.
- S. Knippenberg, D. R. Rehn, M. Wormit, J. H. Starcke, I. L. Rusakova, A. B. Trofimov and A. Dreuw, J. Chem. Phys., 2012, 136, 064107 CrossRef CAS PubMed.
- S. Knippenberg, J. H. Starcke, M. Wormit and A. Dreuw, Mol. Phys., 2010, 108, 2801–2813 CrossRef CAS.
- V. A. Rassolov, J. A. Pople, M. A. Ratner and T. L. Windus, J. Chem. Phys., 1998, 109, 1223–1229 CrossRef CAS PubMed.
- R. A. Kendall, T. H. Dunning and R. J. Harrison, J. Chem. Phys., 1992, 96, 6796–6806 CrossRef CAS PubMed.
- A. Rizzo, S. Coriani and K. Ruud, in Computational Strategies for Spectroscopy, John Wiley & Sons, Inc., 2011, pp. 77–135, DOI:10.1002/9781118008720.ch2.
- T. Thirunamachandran and D. P. Craig, Molecular Quantum Electrodynamics, An Introduction to Radiation Molecule Interaction, Dover Publ., Inc., Mineaol, NY, 1998 Search PubMed.
- W. M. McClain, J. Chem. Phys., 1971, 55, 2789–2796 CrossRef PubMed.
- N. Lin, F. Santoro, A. Rizzo, Y. Luo, X. Zhao and V. Barone, J. Phys. Chem. A, 2009, 113, 4198–4207 CrossRef CAS PubMed.
- R. R. Birge and B. M. Pierce, J. Chem. Phys., 1979, 70, 165–178 CrossRef CAS PubMed.
- K. D. Bonin and T. J. McIlrath, J. Opt. Soc. Am. B, 1984, 1, 52–55 CrossRef CAS.
- F. Wu, G. Zhang, W. Tian, W. Chen, G. Zhao, S. Cao and W. Xie, J. Opt. A: Pure Appl. Opt., 2009, 11, 0652061–0652065 CrossRef.
- F. E. Hernandez, K. D. Belfield, I. Cohanoschi, M. Balu and K. J. Schafer, Appl. Opt., 2004, 43, 5394–5398 CrossRef CAS.
- M. A. C. Nascimento, Chem. Phys., 1983, 74, 51–66 CrossRef CAS.
- M. M. Alam, M. Chattopadhyaya, S. Chakrabarti and A. Rizzo, Phys. Chem. Chem. Phys., 2014, 16, 8030–8035 RSC.
- K. Kristensen, J. Kauczor, A. J. Thorvaldsen, P. Jørgensen, T. Kjærgaard and A. Rizzo, J. Chem. Phys., 2011, 134, 214104 CrossRef PubMed.
- K. Nobusada and K. Yabana, Phys. Rev. A: At., Mol., Opt. Phys., 2007, 75, 032518 CrossRef.
- S. Skourtis, D. Beratan, R. Naaman, A. Nitzan and D. Waldeck, Phys. Rev. Lett., 2008, 101, 238103 CrossRef.
- S. R. Marder, C. B. Gorman, F. Meyers, J. W. Perry, G. Bourhill, J. L. Brédas and B. M. Pierce, Science, 1994, 265, 632–635 CAS.
- M. B. Albota, D. Beljonne, J. L. Bredas, J. E. Ehrlich, J. Y. Fu, A. A. Heikal, S. E. Hess, T. Kogej, M. D. Levin, S. R. Marder, D. McCord-Maughon, J. W. Perry, H. Rockel, M. Rumi, C. Subramaniam, W. W. Webb, X. L. Wu and C. Xu, Science, 1998, 281, 1653–1656 CrossRef CAS.
Footnote |
| † Electronic supplementary information (ESI) available. See DOI: 10.1039/c4ra16732e |
| This journal is © The Royal Society of Chemistry 2015 |