
Synthesis and structures of N-arylcyano-β-diketiminate zinc complexes and adducts and their application in ring‐opening polymerization of l-lactide†
Abstract
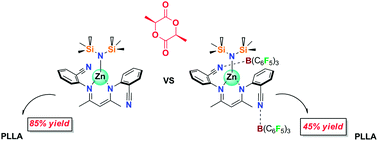
Zinc amide complexes ZnL1N(SiMe3)2, ZnL2N(SiMe3)2 (1 and 2), their tris(pentafluorophenyl)borane adducts ZnL1N(SiMe3)2·B(C6F5)3 (3), ZnL2N(SiMe3)2·2B(C6F5)3 (4), pentafluorophenyl zinc complex ZnL1C6F5 (5) and its adduct ZnL1C6F5·B(C6F5) (6) supported by N-arylcyano-β-diketiminate ligands, as well as bis-ligated Zn(L2)2 (7) were synthesized and characterized by NMR, IR, elemental analysis and X-ray diffraction. Zinc crystal structures of 1, 4, and 7 showed mononuclear complexes, while 2 and 5 were dimmers. ROP of L-lactide with zinc complexes and their B(C6F5)3 adducts leads to generation of poly(L-LA) with high molecular weight and relatively narrow molecular weight distribution. The monomer conversion reached completion in 40 min only for zinc amide complex 1, while for other compounds it was necessary to use at least 5 hours to achieve significant polymerization yields. Coordination of the B(C6F5)3 molecule close to the metal center blocks L-lactide insertion and thus decreases the activity of respective adducts in comparison with borane-free zinc complexes.
 Please wait while we load your content...
Please wait while we load your content...

