
Stereochemistry of cleistanthane diterpenoid glucosides from Phyllanthus emblica†
Abstract
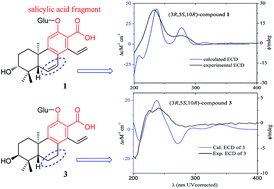
Time-dependent density functional theory (TDDFT) calculated electronic circular dichroism (ECD) and Mosher’s method were applied to establish the absolute configurations of six new highly oxygenated cleistanthane diterpenoid glucosides, phyllanembloids A–F (1–6), isolated from the roots of Phyllanthus emblica. Compounds 1–5 featured a carboxyl group at C-13 adjacent to the hydroxyl group at C-12, and are the first examples of cleistanthane diterpenoids with a salicylic acid fragment. The carboxyl group at C-13 can significantly affect the CD spectrum of cleistanthane diterpenoids, and make the excitations of phenylethylene dominate the Cotton effects (240 and 280 nm), rather than the benzene ring excitations dominating the Cotton effects at 220 and 240 nm as in the 13-methyl analogues. The characteristic Cotton effects of cleistanthane diterpenoids were summarized according to the experimental and calculated ECD curves, which can be applied as a common method to determine the absolute configurations of this type of diterpenoid.
 Please wait while we load your content...
Please wait while we load your content...

