DOI:
10.1039/C4RA16599C
(Paper)
RSC Adv., 2015,
5, 18087-18091
Luminescent MOF material based on cadmium(II) and mixed ligands: application for sensing volatile organic solvent molecules†
Received
18th December 2014
, Accepted 2nd February 2015
First published on 2nd February 2015
Abstract
A luminescent metal–organic framework, [Cd2(TBA)2(bipy)(DMA)2] (1), has been synthesized under solvothermal conditions by employing mixed ligands of 4-(1H-tetrazol-5-yl)-benzoic acid (H2TBA) and 4,4′-bipyridine (bipy). Structure analysis shows that compound 1 is a three-dimensional network with a new (3,4,4)-connected topology. Compound 1 possesses the advantages of good water stability and exceptional thermal stability; it can retain framework crystallinity when it was suspended in water vapor for 10 hours or heated in air at 320 °C. Interestingly, when compound 1 detected different volatile organic solvent molecules, the variation of luminescence intensity depends on the various organic solvents and acetone shows the best quenching behavior. The results indicate that compound 1 may be considered as a potential luminescent probe for the detection of acetone.
Introduction
Metal–organic frameworks (MOFs), as an active research field of functional materials, have drawn considerable attention because of their potential applications in gas storage and separation, catalysis, magnetism, ion exchange, optical and luminescent properties.1–7 In recent years, owing to possessing excellent sensitivity, short response time, simplicity and low cost, luminescent sensing has motivated researcher's interest.8 As one of the most important detection means, it has been extensively used in chemical and biological fields. In fact, luminescent MOFs, with high stability and specific structure characteristic, have been testified promising luminescent sensing materials. There are two main types of sensing applications in chemistry.9 On the one hand, the differential recognition capability of guest molecules can emerge externally optical signals.10 During the activation/de-solvation, the formed open metal sites (OMSs) have the specific ability of coordination or storage volatile organic solvent molecules (VOSMs), which play a crucial role in luminescent response processes. On the other hand, luminescent sensing approach to probe metal ions is very favorable, especially in the field of biological systems and medical science.11 Stimulatingly, luminescent MOFs act as sensing materials have been realized and reported by many research groups, such as Banglin Chen reported a microporous MOF [Zn4(OH)2(1,2,4-BTC)2], which exhibits highly selective sensing of guest molecules and poisonous nitrobenzene,12 and Zhong-Ming Sun stated the luminescent properties of the two microporous Cd-MOFs, which demonstrate the luminescent intensity depends on solvent molecules, and unique selectivity for acetone via a luminescent quenching mechanism.13 Kitagawa and Li utilize the charge transfer electron transitions between the microporous MOFs and the guest substrate molecules for the sensing of aromatic compounds.14 Inspired by the pioneering works on luminescent MOFs sensing, herein, we report a new luminescent MOF [Cd2(TBA)2(bipy)(DMA)2] (1) (H2TBA = 4-(1H-tetrazol-5-yl)-benzoic acid, bipy = 4,4′-bipyridine) with high thermal stability. It is worth pointing out that the sensitive luminescent phenomena were observed when compound 1 exposed to different VOSMs. Meanwhile, the structures, infrared spectra (IR), elemental analyses, powder X-ray diffraction (PXRD), luminescent properties, and thermogravimetric analyses (TGA) were explored in detail.
Experimental
Materials and methods
All chemicals were obtained from commercial sources and used without further purification. Powder X-ray diffraction (XRD) data were collected on a Rigaku D/max-2550 diffractometer with Cu Kα radiation (λ = 1.5418 Å). Elemental analyses (C, H, and N) were achieved by vario MICRO (Elementar, Germany). Infrared (IR) spectra were recorded within a 400–4000 cm−1 region on a Nicolet Impact 410 FTIR spectrometer with KBr pellets. The thermal gravimetric analyses (TGA) were performed on TGA Q500 thermogravimetric analyzer used in air with a heating rate of 10 °C min−1. Fluorescence spectra were collected on a Fluoromax-4 spectrophotometer for the solid powder samples and 1-solvent samples at ambient temperature with the slit width and filter of 3 nm and 450 nm, respectively.
Synthesis of 1
A mixture of Cd(NO3)2·4H2O (0.0061 g, 0.02 mmol), H2TBA, (0.0076 g, 0.04 mmol), 4,4′-bipy (0.0038 g, 0.024 mmol), DMA (1 mL), MeOH (1 mL), HNO3 (100 μL) (2.2 mL HNO3 in 10 mL DMA) was sealed in a 20 mL vial and heated at 85 °C for 36 h, and then cooled to room temperature. The yellow crystals were collected and air-dried (60% yield based on Cd(NO3)2·4H2O). Elemental analysis (%) calc. for C34H34Cd2N12O6, C 43.69, H 3.78, N 17.93; found: C 43.8, H 3.6, N 18.0. The experimental powder X-ray diffraction (XRD) pattern of compound 1 agrees well with the simulated one based on the single-crystal X-ray data, indicating that compound 1 is a pure phase (ESI Fig. S1†).
X-ray crystallography
Crystallographic data for 1 was collected on a Bruker Apex II CCD diffractometer using graphite-monochromated Mo-Kα (λ = 0.71073 Å) radiation at room temperature. The structures were solved by direct methods and refined by full-matrix least-squares on F2 using version 5.1.15 All the metal atoms were located first, and then the oxygen and carbon atoms of the compound were subsequently found in difference Fourier maps. The hydrogen atoms of the ligand were placed geometrically. All non-hydrogen atoms were refined anisotropically. The final formula was derived from crystallographic data combined with elemental and thermogravimetric analysis data. The detailed crystallographic data and selected bond lengths and angles for compound 1 are listed in Tables 1 and S2,† respectively. ESI.† Topology information for 1 was calculated by TOPOS 4.0.16
Table 1 Crystal data and structure refinement for 1
| Compound |
1 |
| Formula |
C34H34Cd2N12O6 |
| fw |
931.53 |
| Temp (K) |
293(2) |
| Crystal system |
Orthorhombic |
| Space group |
Pnna |
| a (Å) |
23.984(5) |
| b (Å) |
13.122(3) |
| c (Å) |
11.917(2) |
| α (°) |
90 |
| β (°) |
90 |
| Γ (°) |
90 |
| V (Å3) |
3750.3(13) |
| Z |
4 |
| Dc (Mg m−3) |
1.650 |
| Absorption coefficient (mm−1) |
1.195 |
| F(000) |
1864 |
| Reflections collected/unique (Rint) |
34![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) 525/4263 [R(int) = 0.0840] 525/4263 [R(int) = 0.0840] |
| Goodness on fit |
1.041 |
| Final R indices [I > 2σ(I)] |
R1 = 0.0401, wR2 = 0.0844 |
| R indices (all data) |
R1 = 0.0640, wR2 = 0.0919 |
| |
|
Results and discussion
Structure description of compound 1
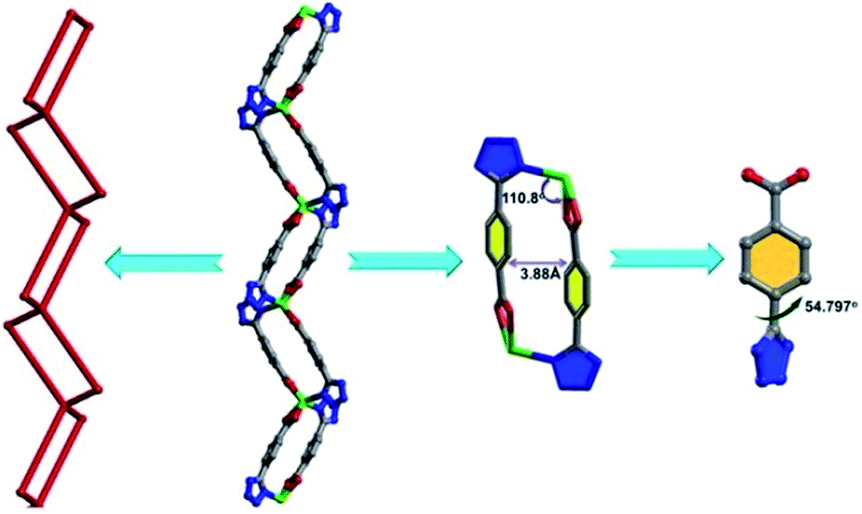
Single-crystal X-ray diffraction analysis reveals that 1 crystallizes in the orthorhombic crystal system with space group of Pnna. In the crystal structure of 1, there are two types of Cd ions. The Cd1 ion adopts an octahedral [CdO2N4] geometry with two nitrogen atoms from the tetrazolate group of two TBA2− ligands and the other two nitrogen atoms from the two bipy ligands located at the equatorial positions, and two oxygen atoms from the coordinated DMA molecules occupied the axial position. The Cd2 ion links with four TBA2− ligands through O,O′-chelating and monodentate N-donor modes to generate a distorted octahedral [CdO4N2] geometry (Fig. S2†). Two TBA2− ligands bridge adjacent Cd2 ions in O,O′-chelating and monodentate N-donor modes, resulting in the formation of a metal–organic square (MOS) with the edge distances of 3.88 and 8.4 Å and vertex angles of 110.8° and 124.8°, respectively. The dihedral angle of the tetrazole and benzene ring is 54.797°.
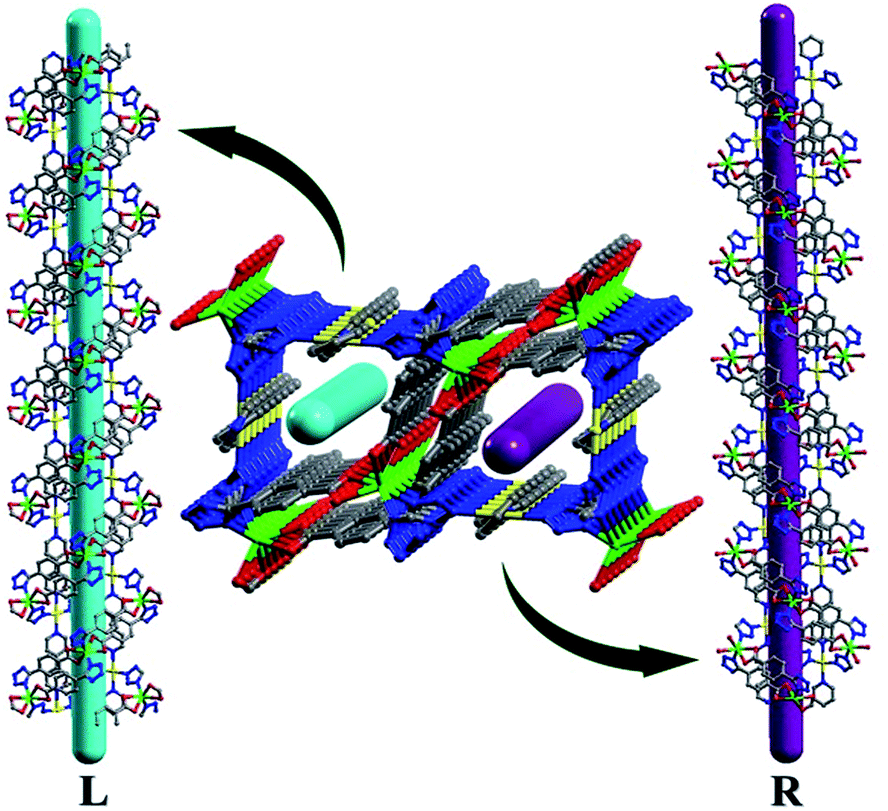
Each MOS interconnects via sharing the Cd2 vertex to give rise to a zigzag chain structure as shown in Fig. 1. And each Cd1 ion connects two bipy molecules to compose a 1D linear chain along the b axis. These zigzag chains which propagated in different directions are alternately connected by linear chains to afford a 3D framework (Fig. 2). The Cd1–O bond distance is 2.332(3) Å, slightly shorter than that of Cd2–O with the range Of 2.349(3)–2.368(3) Å, and Cd–N bond distances are in the range of 2.298(3)–2.412(4) Å, respectively, which are close to those reported in Cd-MOF compounds. Additionally, it is interesting to note that compound 1 exhibits two types of helical chains with opposite helical directions (left- and right-handed) running along the c axis with a pitch of 11.92 Å (Fig. 3 and S3†).
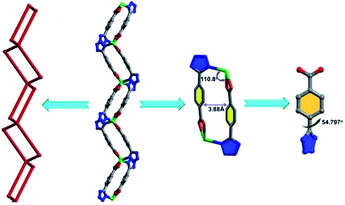 |
| | Fig. 1 The metal–organic square (MOS) view and the dihedral angles between phenyl and tetrazole rings. | |
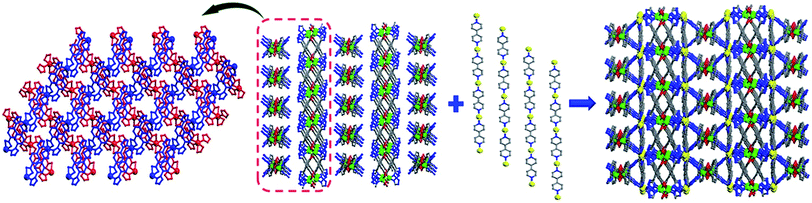 |
| | Fig. 2 3D framework constructed by different directional zigzag chains and linear chains. | |
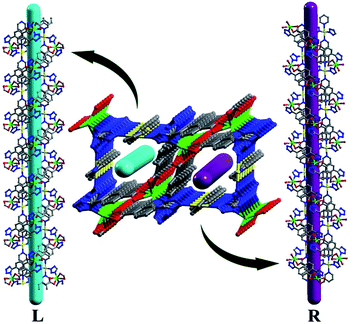 |
| | Fig. 3 View of helixes with opposite handedness. | |
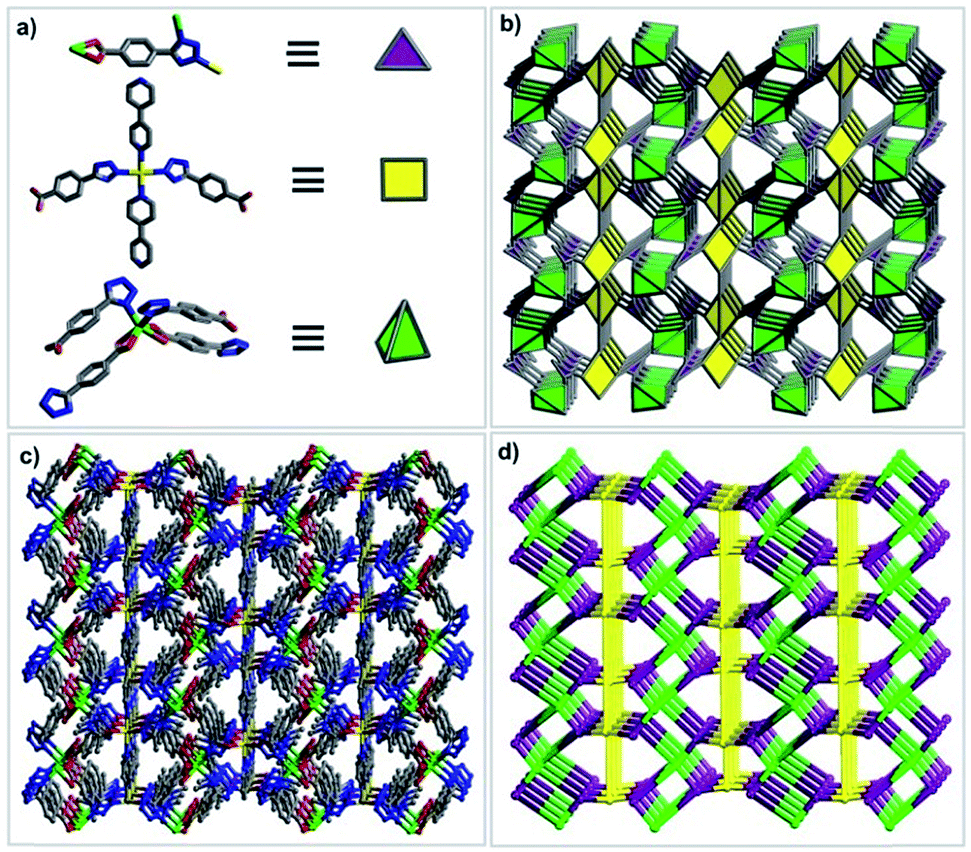
In order to illustrate the structure of compound 1 more clearly, the network topology of compound 1 is further analyzed. The TBA2− ligands act as a 3-connected nodes linking to two Cd2 atoms and one Cd1 atom, the bipy ligands can be viewed as a bridging linker between two Cd1 nodes, and the metal centers can be regarded as 4-connected nodes.
The Cd1 atom coordinated with two bipy ligands and two TBA2− ligands can be viewed as a square node, the Cd2 atoms linked with four TBA2− ligands to give rise to a tetrahedral node. As a result, the topology of compound 1 can be simplified into a (3,4,4)-connected net, which represents a new network topology (Fig. 4). Overall, compound 1 is assembled by the ternary building units (one kind of organic secondary building unit (SBU) and two kinds of inorganic SBUs): triangle, square and tetrahedron (Table S1†).
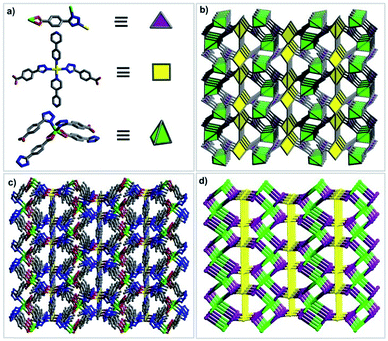 |
| | Fig. 4 Description of the structure of 1: (a) ternary SBUs; (b) polyhedral view of the net; (c) ball and stick model of the 3D framework; (d) a schematic representation of the new net. | |
Thermogravimetric analysis
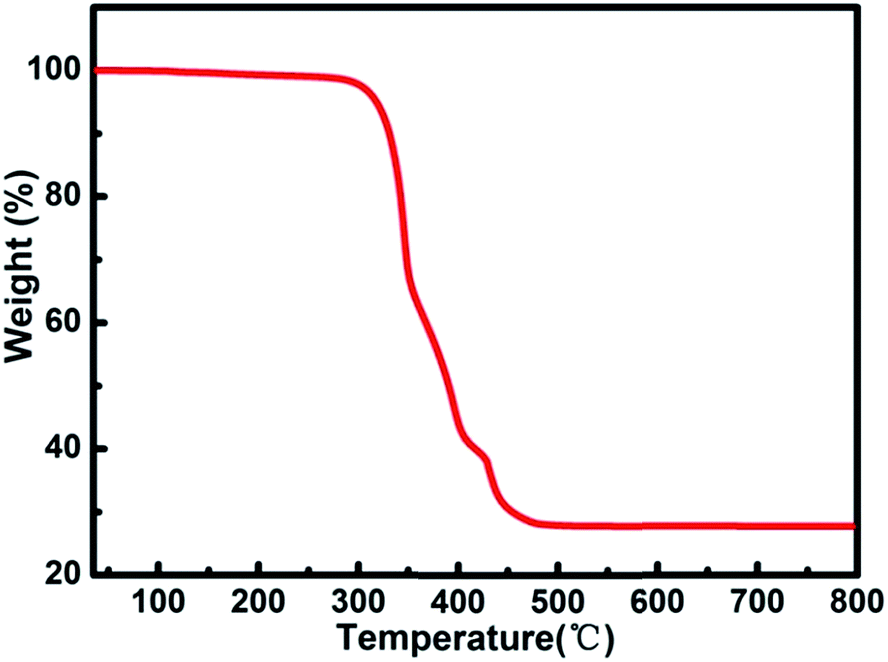
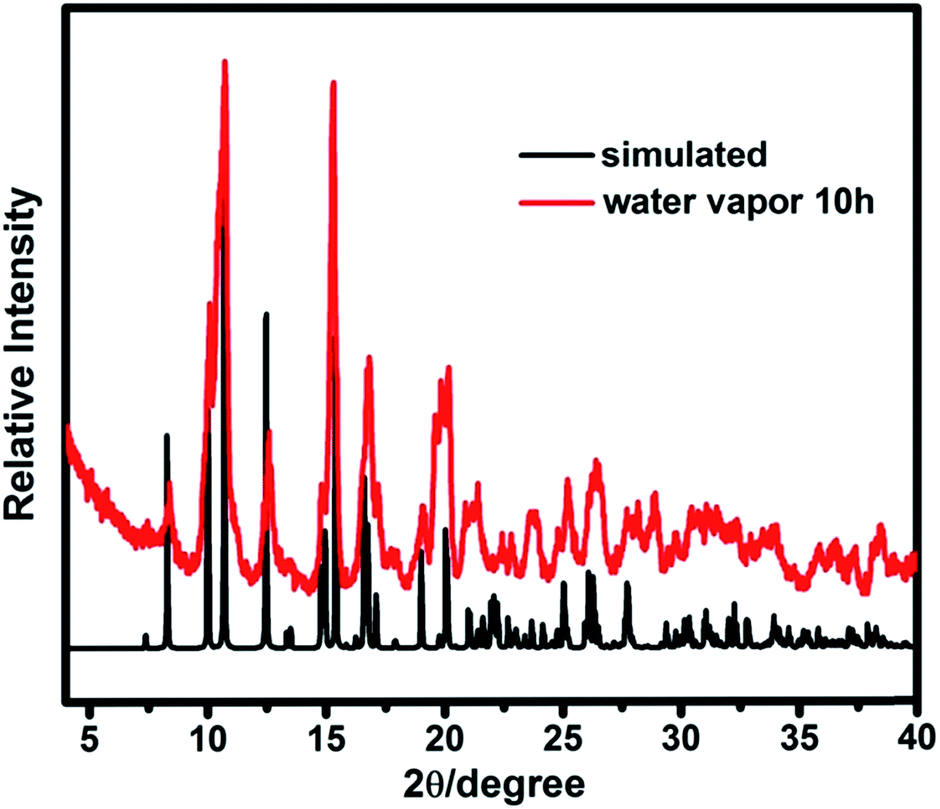
Thermogravimetric (TG) analysis measurement indicated that compound 1 exhibits only one step weight loss of 72.8% (calc: 73.4%) between 330 and 500 °C, which is assigned to the departure of the coordinated DMA molecules together with the structure collapse. The pore windows of compound 1 are too small and prevent the coordinated DMA molecules going out (Fig. 5). In order to demonstrate the thermal stability of compound 1, its temperature-dependent powder XRD (Fig. S5†) are recorded, which is accordant with the inflection points of TGA curve. The results show that the framework of compound 1 is thermally stable up to 320 °C. Powder XRD studies indicate that the final product, upon calcinations above 600 °C, is a main phase of CdO (JCPDS: 05-0640) (Fig. S6†). The as-synthesized compound 1 shows good stability in the water vapor, which can be suspended in water vapor for 10 h without losing crystallinity (Fig. 6).
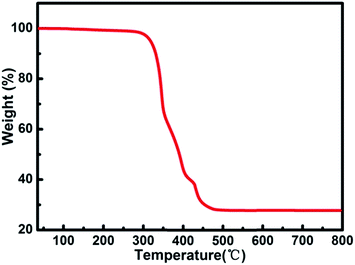 |
| | Fig. 5 TG analysis curve of compound 1. | |
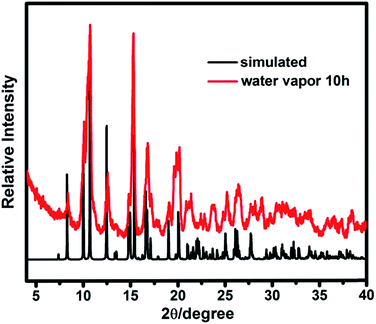 |
| | Fig. 6 The power XRD patterns of compound 1 for the calculated and suspended in water vapor. | |
Luminescent properties
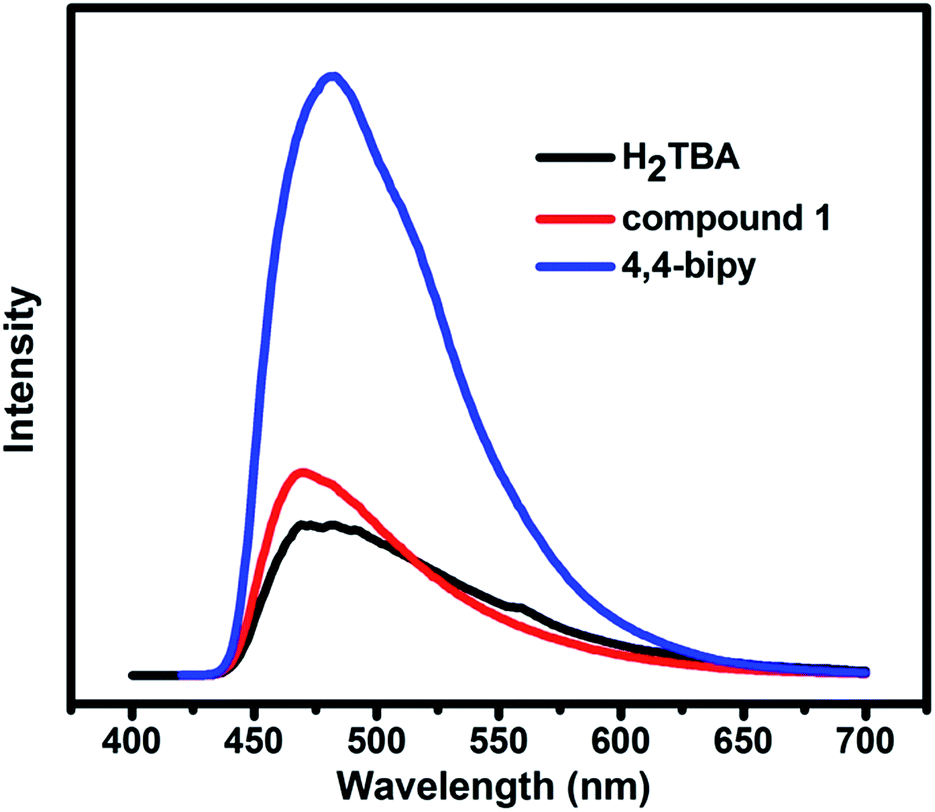
MOFs constructed by d10 metal ions with excellent luminescent property have the potential to become promising candidates for luminescent materials.17 Therefore, the solid-state luminescent properties of free ligands H2TBA, bipy, and compound 1 were evaluated at ambient temperature (Fig. 7). The free ligands are observed with emission maxima at 480 nm (λex = 310 nm) for H2TBA and 480 nm (λex = 400 nm) for bipy, respectively. Compound 1 exhibits the same emission characteristic as the ligands, and the emission peak is 470 nm (λex = 400 nm). The luminescence emission can probably be contributed to the influence of two kinds of ligands with metal centers.18
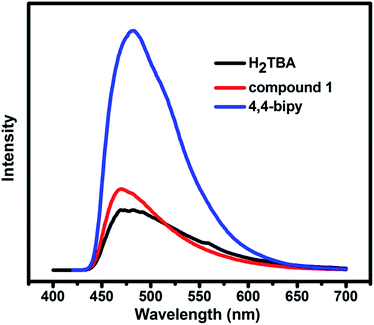 |
| | Fig. 7 Solid-state emission spectra of H2TBA, 4,4-bipy and the compound 1. | |
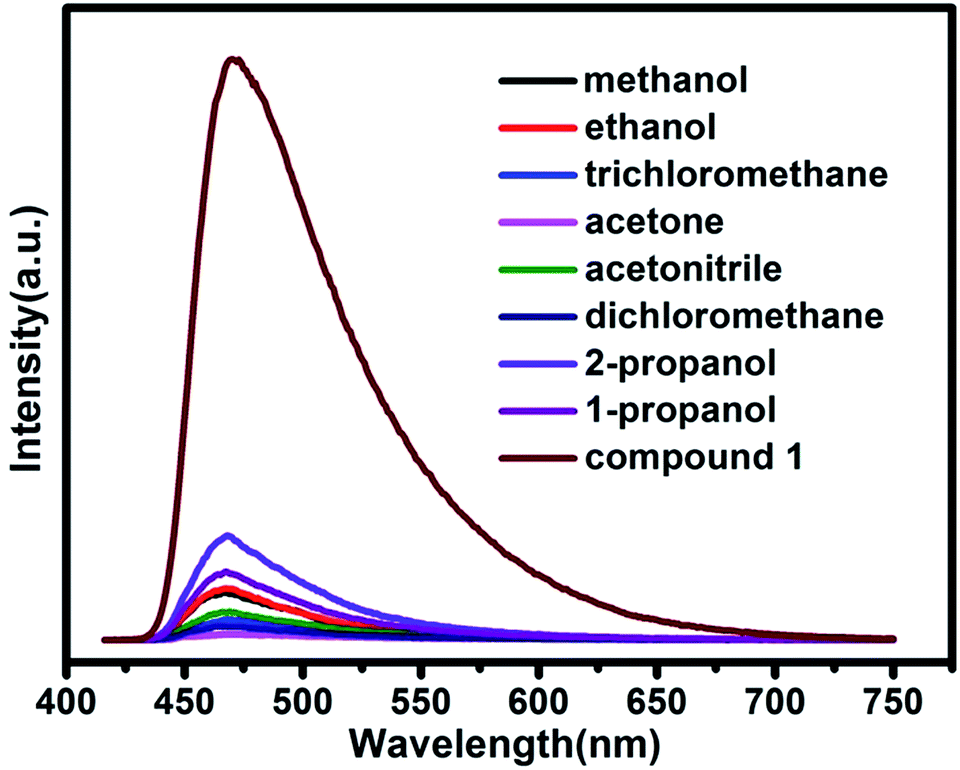
Further luminescence sensing measurements were performed to investigate the influence of various guest molecules towards compound 1. The luminescent spectra of compound 1 in various solvent emulsions at ambient temperature were investigated. The 1-solvent emulsions were prepared by introducing 3 mg powder sample of compound 1 into 3.0 mL of methanol (MeOH), ethanol (EtOH), 1-propanol (1-PA), 2-propanol (2-PA), acetone (CH3COCH3), acetonitrile (CH3CN), dichloromethane (CH2Cl2), trichloromethane (CHCl3), and were then vigorously agitated by using ultrasound to form stable emulsions before fluorescence study. As shown in Fig. 8, the luminescent spectra of various solvent emulsions are similar to that of compound 1 when excitation at 400 nm. The most remarkable feature is that the luminescent intensity significantly changes with different organic solvents, particularly in the case of acetone, which exhibits the most quenching behavior. The order of decreasing efficiency is CH3COCH3 > CH2Cl2 > CHCl3 > CH3CN > MeOH > EtOH > 1-PA > 2-PA. The results demonstrate that compound 1 may be used as a promising material for detection of acetone, corresponding to the reported Cd-MOF materials.13,19 The quenching behavior of the acetone might be ascribed to the interaction between “C![[double bond, length as m-dash]](https://www.rsc.org/images/entities/char_e001.gif) O” of acetone and the framework of compound 1. Although the mechanism of response to organic solvents is still not clear at this moment, the interaction of the crystals and solvent plays an important role in such solvent-dependent luminescent property.
O” of acetone and the framework of compound 1. Although the mechanism of response to organic solvents is still not clear at this moment, the interaction of the crystals and solvent plays an important role in such solvent-dependent luminescent property.
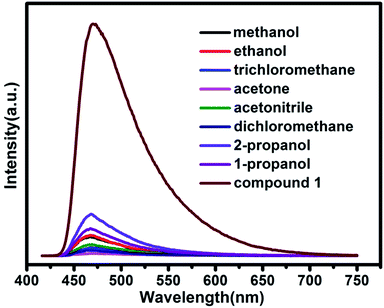 |
| | Fig. 8 Emission spectra of compound 1 in different volatile organic solvents. | |
Conclusions
In summary, by utilizing the mixed ligands of H2TBA and bipy, we have successfully assembled a 3D luminescent MOF material with (3,4,4)-connected network topology. Compound 1 exhibits exceptional thermal stability and good water stability. Luminescent property of compound 1 in various VOSMs indicates that it may be used as a potential luminescent probe for the detection of acetone. This work may enrich the family of luminescent MOFs chemistry and expand the potential applications of such materials.
Acknowledgements
The authors gratefully acknowledge the financial support of the Natural Science Foundation of China (Grant nos 21373095, 21371067 and 21171064).
Notes and references
-
(a) P. Nugent, Y. Belmabkhout, S. D. Burd, A. J. Cairns, R. Luebke, K. Forrest, T. Pham, S. Ma, B. Space, L. Wojtas, M. Eddaoudi and M. J. Zaworotko, Nature, 2013, 495, 80–84 CrossRef CAS PubMed;
(b) M. P. Suh, H. J. Park, T. K. Prasad and D. W. Lim, Chem. Rev., 2012, 112, 782–835 CrossRef CAS PubMed;
(c) A. Phan, C. J. Doonan, F. J. Uribe-Romo, C. B. Knobler, M. O'Keeffe and O. M. Yaghi, Acc. Chem. Res., 2010, 43, 58–67 CrossRef CAS PubMed;
(d) K. Sumida, D. L. Rogow, J. A. Mason, T. M. McDonald, E. D. Bloch, Z. R. Herm, T. H. Bae and J. R. Long, Chem. Rev., 2012, 112, 724–781 CrossRef CAS PubMed;
(e) J. R. Li, J. Sculley and H. C. Zhou, Chem. Rev., 2012, 112, 869–932 CrossRef CAS PubMed;
(f) P. Z. Li, X. J. Wang, K. Zhang, A. Nalaparaju, R. Zou, J. Jiang and Y. Zhao, Chem. Commun., 2014, 50, 4683–4685 RSC.
-
(a) W. Y. Gao, Y. Chen, Y. Niu, K. Williams, L. Cash, P. J. Perez, L. Wojtas, J. Cai, Y. S. Chen and S. Ma, Angew. Chem., Int. Ed., 2014, 53, 2615–2619 CrossRef CAS PubMed;
(b) M. Yoon, R. Srirambalaji and K. Kim, Chem. Rev., 2012, 112, 1196–1231 CrossRef CAS PubMed;
(c) J. Lee, O. K. Farha, J. Roberts, K. A. Scheidt, S. T. Nguyen and J. T. Hupp, Chem. Soc. Rev., 2009, 38, 1450–1459 RSC;
(d) D. Wang, T. Zhao, Y. Cao, S. Yao, G. Li, Q. Huo and Y. Liu, Chem. Commun., 2014, 50, 8648–8650 RSC;
(e) P. Z. Li, X. J. Wang, R. H. D. Tan, Q. Zhang, R. Zou and Y. Zhao, RSC Adv., 2013, 3, 15566–15570 RSC.
-
(a) W. Zhang and R. G. Xiong, Chem. Rev., 2012, 112, 1163–1195 CrossRef CAS PubMed;
(b) L. Wang, J. Morales, T. Wu, X. Zhao, W. P. Beyermann, X. Bu and P. Feng, Chem. Commun., 2012, 48, 7498–7500 RSC;
(c) Y. Q. Wang, Q. Yue, Y. Qi, K. Wang, Q. Sun and E. Q. Gao, Inorg. Chem., 2013, 52, 4259–4268 CrossRef CAS PubMed;
(d) L. Lili, Z. Xin, R. Shumin, Y. Ying, D. Xiaoping, G. Jinsen, X. Chunming and H. Jing, RSC Adv., 2014, 4, 13093–13107 RSC.
-
(a) S. Das, H. Kim and K. Kim, J. Am. Chem. Soc., 2009, 131, 3814–3815 CrossRef CAS PubMed;
(b) F. Nouar, J. Eckert, J. F. Eubank and P. Forster, J. Am. Chem. Soc., 2009, 131, 2864–2870 CrossRef CAS PubMed;
(c) H. Y. Lin, J. Luan, X. L. Wang, J. W. Zhang, G. C. Liu and A. X. Tian, RSC Adv., 2014, 4, 62430–62445 RSC.
- C. Wang, T. Zhang and W. Lin, Chem. Rev., 2012, 112, 1084–1104 CrossRef CAS PubMed.
-
(a) L. E. Kreno, K. Leong, O. K. Farha, M. Allendorf, R. P. Van Duyne and J. T. Hupp, Chem. Rev., 2012, 112, 1105–1125 CrossRef CAS PubMed;
(b) M. E. Germain and M. J. Knapp, Chem. Soc. Rev., 2009, 38, 2543–2555 RSC;
(c) W. Zhang, H. Huang, D. Liu, Q. Yang, Y. Xiao, Q. Ma and C. Zhong, Microporous Mesoporous Mater., 2013, 171, 118–124 CrossRef CAS PubMed;
(d) J. Ferrando-Soria, H. Khajavi, P. Serra-Crespo, J. Gascon, F. Kapteijn, M. Julve, F. Lloret, J. Pasán, C. Ruiz-Pérez, Y. Journaux and E. Pardo, Adv. Mater., 2012, 24, 5625–5629 CrossRef CAS PubMed;
(e) B. Chen, Y. Yang, F. Zapata, G. Lin, G. Qian and E. B. Lobkovsky, Adv. Mater., 2007, 19, 1693–1696 CrossRef CAS.
-
(a) Y. Cui, Y. Yue, G. Qian and B. Chen, Chem. Rev., 2012, 112, 1126–1162 CrossRef CAS PubMed;
(b) D. Ma, B. Li, X. Zhou, Q. Zhou, K. Liu, G. Zeng, G. Li, Z. Shi and S. Feng, Chem. Commun., 2013, 49, 8964–8966 RSC;
(c) B. Xiao, Q. Zhang, C. Huang and Y. Li, RSC Adv., 2015, 5, 2857–2860 RSC;
(d) W. Sun, J. Wang, G. Zhang and Z. Liu, RSC Adv., 2014, 4, 55252–55255 RSC;
(e) H. Zhang, P. Lin, X. Shan, F. Du, Q. Li and S. Du, Chem. Commun., 2013, 49, 2231–2233 RSC;
(f) Q. Li and S. Du, RSC Adv., 2015, 5, 9898–9903 RSC;
(g) Q. Li and S. Du, RSC Adv., 2014, 4, 30963–30967 RSC.
-
(a) L. Basabe-Desmonts, D. N. Reinhoudt and M. Crego-Calama, Chem. Soc. Rev., 2007, 36, 993–1017 RSC;
(b) A. J. Lan, K. H. Li, H. H. Wu, D. H. Olson, T. J. Emge, W. Ki, M. C. Hong and J. Li, Angew. Chem., Int. Ed., 2009, 48, 2334–2338 CrossRef CAS PubMed.
-
(a) S. Liu, Z. Xiang, Z. Hu, X. Zheng and D. Cao, J. Mater. Chem., 2011, 21, 6649–6653 RSC;
(b) Q. K. Liu, J. P. Ma and Y. B. Dong, Chem. Commun., 2011, 47, 7185–7187 RSC;
(c) C. Zhang, Y. Che, Z. Zhang, X. Yang and L. Zang, Chem. Commun., 2011, 47, 2336–2338 RSC.
-
(a) B. Chen, S. Xiang and G. Qian, Acc. Chem. Res., 2010, 43, 1115–1124 CrossRef CAS PubMed;
(b) S. Pramanik, C. Zheng, X. Zhang, T. J. Emge and J. Li, J. Am. Chem. Soc., 2011, 133, 4153–4155 CrossRef CAS PubMed.
- Y. Xiao, Y. Cui, Q. Zheng, S. Xiang, G. Qian and B. Chen, Chem. Commun., 2010, 46, 5503–5505 RSC.
- Z. Zhang, S. Xiang, X. Rao, Q. Zheng, F. R. Fronczek, G. Qian and B. Chen, Chem. Commun., 2010, 46, 7205–7207 RSC.
- F. Y. Yi, W. T. Yang and Z. M. Sun, J. Mater. Chem., 2012, 22, 23201–23209 RSC.
-
(a) A. Lan, K. Li, H. Wu, D. H. Olson, T. J. Emge, W. Ki, M. Hong and J. Li, Angew. Chem., Int. Ed., 2009, 48, 2334–2338 CrossRef CAS PubMed;
(b) D. Tanaka, S. Horike, S. Kitagawa, M. Ohba, M. Hasegawa, Y. Ozawac and K. Toriumi, Chem. Commun., 2007, 3142–3144 RSC.
- G. M. Sheldrick, SHELXTL-97, Program for Crystal Structure Refinement, University of Gottingen, 1997 Search PubMed.
- V. A. Blatov, A. P. Shevchenko and D. M. Proserpio, Cryst. Growth Des., 2014, 14, 3576–3683 CAS.
-
(a) X. Chen, Y. Tong, M. M. Han, K. L. Cao and Y. L. Feng, Inorg. Chem. Commun., 2014, 40, 62–65 CrossRef CAS PubMed;
(b) D. Mal, R. Sen, P. Brandao, F. Shi, R. A. S. Ferreira and Z. Lin, Inorg. Chem. Commun., 2014, 40, 92–96 CrossRef CAS PubMed.
- Q. Li and J. Qian, RSC Adv., 2014, 4, 32391–32397 RSC.
- J. M. Zhou, W. Shi, H. M. Li, H. Li and P. Cheng, J. Phys. Chem. C, 2013, 118, 416–426 Search PubMed.
Footnote |
| † Electronic supplementary information (ESI) available: Selected bond lengths and angles, topology information, additional figures for crystal structures, PXRD and IR. CCDC 1031697. For ESI and crystallographic data in CIF or other electronic format see DOI: 10.1039/c4ra16599c |
|
| This journal is © The Royal Society of Chemistry 2015 |
Click here to see how this site uses Cookies. View our privacy policy here.