
Antineoplastic agents 596. Isolation and structure of chromomycin A5 from a Beaufort Sea microorganism†‡
Abstract
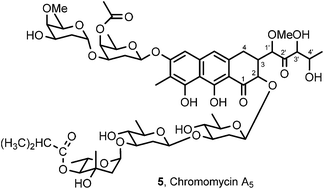
A microorganism identified as Streptomyces sp. was isolated from a sedimentary specimen collected on the Beaufort Sea coast of Alaska. Fermentation scale-up and cancer cell line-guided separation of the constituents led to isolation and structural elucidation (high resolution MALDI-TOF mass and 2D NMR spectral analyses) of a diastereomer of chromomycin A2 (2) designated chromomycin A5 (5). Chromomycin A5 displayed very potent cancer cell growth inhibitory activity (reaching with MCF-7 breast cancer GI50 0.00073 μg mL−1) against a mini-panel of murine and human cancer cell lines.
 Please wait while we load your content...
Please wait while we load your content...

