
Synthesis of fluorovinyl aryl ethers by a three-component reaction of gem-difluoroalkenes with arylboronic acids and oxygen†
Mingjin Wang,
Fang Liang,
Yang Xiong and
Song Cao*
Shanghai Key Laboratory of Chemical Biology, School of Pharmacy, East China University of Science and Technology (ECUST), Shanghai 200237, China. E-mail: scao@ecust.edu.cn; Fax: +86-21-64252603; Tel: +86-21-64253452
First published on 14th January 2015
Abstract
A novel and efficient three-component reaction for the synthesis of fluorovinyl aryl ethers from gem-difluoroalkenes, arylboronic acids and oxygen under metal-free conditions is described.
Vinyl ethers or fluorovinyl ethers are often found as structural frameworks in material chemistry and medicinal chemistry.1 They also serve as versatile building blocks and intermediates in organic synthesis for the preparation of complex molecules.2 A facile method for the synthesis of vinyl ethers is transition metal catalyzed cross-coupling of vinyl halides (RX, X = Cl, Br, and I) and phenols (Scheme 1a).3 More recently, Kawatsura reported an efficient protocol for the synthesis of vinyl aryl ether catalyzed by Pd(PPh3)4 via carbon–fluorine bond activation (Scheme 1b).4
 | ||
| Scheme 1 Synthesis of vinyl aryl ethers or fluorovinyl aryl ethers. | ||
gem-Difluoroalkenes possess remarkable reactivity toward nucleophiles due to the high polarization of the carbon–carbon double bond caused by the two fluorine atoms.5 Thus, a straightforward method for the synthesis of fluorovinyl aryl ethers is by the reaction of gem-difluoroalkenes with phenols in presence of base via nucleophilic substitution of the vinylic fluorine (Scheme 1c).6 However, the major drawback of this approach is the formation of the undesired addition or disubstitution byproducts.7 Therefore, the development of a facile and efficient approach for the synthesis of fluorovinyl aryl ethers from gem-difluoroalkenes is still highly desirable. In continuation of our efforts on the functionalization of carbon–fluorine bonds in gem-difluoroalkenes,8 we herein describe a new and facile approach to fluorovinyl aryl ethers through a three-component reaction of gem-difluoroalkenes, arylboronic acids and oxygen with the assistance of K3PO4 (Scheme 1d).
It is well-known that the hydroxylation of arylboronic acids to phenols usually occurred readily under various reaction conditions.9 In addition, the formation of phenols from the corresponding arylboronic acids was reported as side reaction of the palladium-catalyzed Suzuki–Miyaura cross-coupling reaction.10 In our recent publication, we have reported the synthesis of polyfluoro-substituted unsymmetrical biaryl ethers via a novel Ni-catalyzed reaction of polyfluoroarenes with arylboronic acids and oxygen.11 Encouraged by the results obtained with the polyfluoroarenes, we wondered if this strategy could be further extended into the formation of fluorovinyl aryl ether from gem-difluoroalkenes. To test our assumption, the investigation of the reaction conditions was commenced using (2,2-difluoroethene-1,1-diyl)dibenzene 1a and phenylboronic acid 2a as the model substrates, as shown in Table 1.
|
||||||
|---|---|---|---|---|---|---|
| Entry | Catalyst (5 mol%) | 2a (equiv.) | Base (2.0 equiv.) | Solvent | Temp (°C) | Yield of 3aab (%) |
| a Reaction conditions: gem-difluoroalkenes 1a (1.0 mmol), solvent (2 mL), 24 h, under air atmosphere.b Yields determined by GC analysis.c Under an argon atmosphere. | ||||||
| 1 | NiCl2 | 2.0 | K3PO4 | THF | 80 | 10 |
| 2 | NiCl2 | 2.0 | K3PO4 | Dioxane | 100 | 33 |
| 3 | NiCl2 | 2.0 | K3PO4 | DMSO | 100 | 36 |
| 4 | NiCl2 | 2.0 | K3PO4 | NMP | 100 | 83 |
| 5 | Ni(acac)2 | 2.0 | K3PO4 | DMSO | 100 | 50 |
| 6 | Ni(acac)2 | 2.0 | K3PO4 | DMF | 100 | 66 |
| 7 | Ni(acac)2 | 2.0 | K3PO4 | NMP | 100 | 92 |
| 8 | None | 2.0 | K3PO4 | NMP | 100 | 93 |
| 9 | None | 2.0 | K3PO4 | NMP | 90 | 79 |
| 10 | None | 2.0 | K3PO4 | NMP | 80 | 70 |
| 11 | None | 1.5 | K3PO4 | NMP | 100 | 65 |
| 12 | None | 1.2 | K3PO4 | NMP | 100 | 48 |
| 13 | None | 1.0 | K3PO4 | NMP | 100 | 33 |
| 14 | None | 2.0 | None | NMP | 100 | 0 |
| 15 | None | 2.0 | Et3N | NMP | 100 | 0 |
| 16 | None | 2.0 | Na2CO3 | NMP | 100 | 71 |
| 17 | None | 2.0 | KOH | NMP | 100 | 86 |
| 18 | None | 2.0 | tBuOK | NMP | 100 | 92 |
| 19 | None | 2.0 | Cs2CO3 | NMP | 100 | 91 |
| 20 | Nonec | 2.0 | K3PO4 | NMP | 100 | 48 |
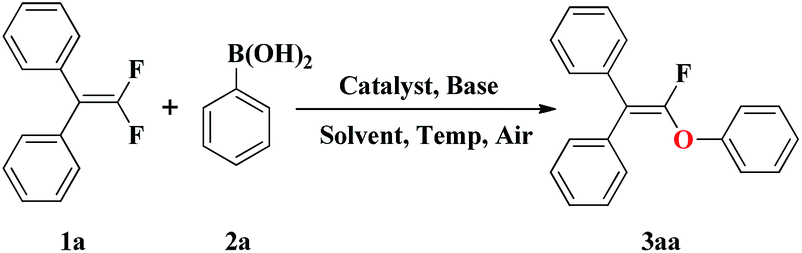
Initially, the reaction was performed in the presence of NiCl2 or Ni(acac)2 using K3PO4 as base. Solvent screening (entries 1–7) showed that aprotic polar solvent NMP were most suitable reaction media while nonpolar solvent, such as THF was not effective. To our delight, the desired product 3aa was obtained in excellent yield using K3PO4 as base and NMP as solvent at 100 °C for 24 h in absence of Ni catalyst (entry 8). Decreasing the reaction temperature from 100 to 80 °C would result in lower yields (entries 9 and 10). The yield of product 3aa was promoted to 93% when the amount of phenylboronic acid 2a was increased to 2.0 equiv. (entries 8, 11–13). Moreover, the use of two equivalent of base was proven to be essential for the success of the transformation (entries 14–19).
No detectable product was observed when performing the reaction in the absence of base (entry 14). Among all the tested bases, K3PO4 provided the most optimal yield of 93% (entry 8). When tBuOK and Cs2CO3 were employed, comparable yields to K3PO4 were obtained (entries 18–19). Finally, when the reaction was performed under argon, only 48% the expected product was produced (entry 20). It indicated that the atmospheric oxygen plays a key role in the reaction. The formation of 3aa (48% yield) under an argon atmosphere is due to the existence of small amounts of oxygen in the reaction system.
To survey the generality of this novel three-component reaction, a number of different symmetrical gem-difluoroalkenes was allowed to react with a wide array of arylboronic acids under the optimized conditions (Table 1, entry 8). The results are elucidated in Table 2. As can be seen from Table 2, in most cases the desired fluorovinyl aryl ethers could be obtained in moderate to good yields. Arylboronic acids bearing electron-withdrawing group (3ag and 3ah) gave the desired products in higher yields than substrates bearing electron-donating groups (3ab and 3ad). The presence of electron-donating group on the benzene ring of symmetrical gem-difluoroalkene was found to be advantageous over other gem-difluoroalkenes (3df versus 3af). Interestingly, arylboronic acid having sensitive functional group such as aldehyde group remains intact under the optimized conditions (3ah).
|
|---|
| a Reaction conditions: gem-difluoroalkenes 1a–d (1.0 mmol), ArB(OH)2 2a–k (2.0 mmol), K3PO4 (2.0 mmol), NMP (2 mL), 24 h, under air atmosphere.b Isolated yields.c Reaction conditions: gem-difluoroalkenes 1e–g (1.0 mmol), PhB(OH)2 2a (2.0 mmol), Ni(acac)2 (5 mol%), Cs2CO3 (2.0 mmol), toluene (2 mL), 24 h, O2 balloon was applied. |
 |

Treatment of the heteroarylboronic acids such as pyridin-3-ylboronic acid 2i, pyridin-4-ylboronic acid 2j and thiophen-3-ylboronic acid 2k with (2,2-difluoroethene-1,1-diyl)dibenzene 1a afforded the corresponding products in good to high yields (3ai–ak). The synthesis of 3ai–ak is particularly useful and could be applied to prepare some special fluorovinyl aryl ethers when the corresponding phenols such as thiophen-3-ol are unstable or unavailable.
The 4,4′-(2,2-difluoroethene-1,1-diyl)bis(chlorobenzene) 1e, 4,4′-(2,2-difluoroethene-1,1-diyl)bis-(bromobenzene) 1f and 9-(difluoromethylene)-9H-fluorene 1g were not good substrates for this reaction under the optimized conditions due to their low reactivities towards ArO−. According to our previous report,11 Ni catalyst could accelerate the releasing of aryloxy anion (ArO−). Therefore, we reinvestigated the reaction conditions. It was found that the addition of 5 mol% Ni(acac)2 could improved the yield obviously. Furthermore, performing the reaction under an oxygen atmosphere (balloon) resulted in a higher yield of the expected product than when the reaction was performed under air (see Table 2, note c).
For the unsymmetrical difluoroalkenes 1h–k (one is hydrogen and the other is aryl group), the reactions also gave the corresponding products 3ha–kl in good to high yields but with low E/Z-selectivity under the optimized conditions. The results are summarized in Table 3. Unfortunately, the E and Z isomers of 3ha–kl are inseparable by column chromatography.
|
|---|
| a Reaction conditions: unsymmetrical difluoroalkenes 1h–k (1.0 mmol), ArB(OH)2 2a, 2d, 2f, 2l (2.0 mmol), K3PO4 (2.0 mmol), NMP (2 mL), 24 h, under air atmosphere.b Isolated yields of an inseparable E/Z mixture of the products.c E/Z selectivity was determined by 19F NMR spectra. The configurations of E- and Z-isomers were determined by their 3JHF coupling constants in 1H NMR spectra. |
 |

On the basis of the experimental results and our previous work,11 we suggest that the mechanism is analogous to those reported by Ichikawa and co-workers,5d gem-difluoroalkenes undergo nucleophilic vinylic substitution (SNV) with aryloxy anion (ArO−) in the presence of K3PO4 via addition–elimination processes to afford fluorovinyl aryl ethers. The oxidation of arylboronic acids with oxygen to generate the corresponding aryloxy anion is essential for the efficient aryloxylation of gem-difluoroalkenes. It is reasonable to assume that the highly electron-deficient gem-difluoroalkenes are more efficient for trapping aryloxy anion generated from arylboronic acid than poly-fluorinated aromatic compounds.
In summary, we have developed an efficient three-component reaction for the preparation of fluorovinyl aryl ethers from gem-difluoroalkenes, arylboronic acids and oxygen using simple and commercially available K3PO4 as base under metal-free conditions. This reaction proceeds well for a wide variety of arylboronic acids including heteroarylboronic acids and tolerates several electron-donating as well as electron-withdrawing functional groups. It provides an alternative approach to access fluorovinyl aryl ethers.
Acknowledgements
We are grateful for financial supports from the National Natural Science Foundation of China (Grant nos 21472043, 21272070), and the Key Project in the National Science & Technology Pillar Program of China in the twelfth five-year plan period (2011BAE06B01-15).Notes and references
- (a) D. K. Dei, B. R. Lund, J.-B. Wu, D. Simon, T. Ware, W. E. Voit, D. MacFarlane, S. M. Liff and D. W. Smith Jr, ACS Macro Lett., 2013, 2, 35 CrossRef CAS; (b) S. Keck, T. A. Knoerzer, D. W. Smith Jr and S. T. Lacono, Polym. Int., 2013, 62, 1485 CrossRef CAS; (c) S.-B. Lee, Y.-J. Kim, U. Ko, C.-M. Min, M.-K. Ahn, S.-J. Chung, I.-S. Moon and J.-S. Lee, J. Membr. Sci., 2014, 456, 49 CrossRef CAS PubMed; (d) C. Yuan, K.-K. Jin, K. Li, S. Diao, J.-W. Tong and Q. Fang, Adv. Mater., 2013, 25, 4875 CrossRef CAS PubMed; (e) E. L. Luzina and A. V. Popov, Eur. J. Med. Chem., 2009, 44, 4944 CrossRef CAS PubMed; (f) J.-K. Kim, D.-H. Kim, H.-H. Kim, K.-H. Kim, C.-S. Yoon and I.-C. Hwang, WO Pat., 2009005297, 2009.
- (a) A. V. Matsnev, S.-Y. Qing, M. A. Stanton, K. A. Berger, G. Haufe and J. S. Thrasher, Org. Lett., 2014, 16, 2402 CrossRef CAS PubMed; (b) G.-L. Lu, S. Zhang, Y.-J. Li and X.-Y. Huang, Chin. J. Chem., 2011, 29, 2791 CrossRef CAS; (c) T. Kita, K. Yokoyama, S. Ihara, T. Asai and H. Koh, WO Pat., 2013031603, 2013; (d) P. G. Wilson, J. M. Percy, J. M. Redmond and A. W. McCarter, J. Org. Chem., 2012, 77, 6384 CrossRef CAS PubMed.
- (a) D. Kundu, P. Maity and B. C. Ranu, Org. Lett., 2014, 16, 1040 CrossRef CAS PubMed; (b) G. Nordmann and S. L. Buchwald, J. Am. Chem. Soc., 2003, 125, 4978 CrossRef CAS PubMed; (c) J. Limberger, B. C. Leal, D. F. Back, J. Dupont and A. L. Monteiro, Adv. Synth. Catal., 2012, 354, 1429 CrossRef CAS; (d) Z.-H. Wan, C. D. Jones, T. M. Koenig, Y. J. Pu and D. Mitchell, Tetrahedron Lett., 2003, 44, 8257 CrossRef CAS PubMed.
- E. Nomada, H. Watanabe, M. Yamamoto, T. Udagawa, B. Zhou, A. Kobayashi, M. Minakawa and M. Kawatsura, Synlett, 2014, 25, 1725 CrossRef PubMed.
- (a) M. S. Kim and I. H. Jeong, Tetrahedron Lett., 2005, 46, 3545 CrossRef CAS PubMed; (b) X.-H. Huang, P.-Y. He and G.-Q. Shi, J. Org. Chem., 2000, 65, 627 CrossRef CAS; (c) T. Fujita, K. Sakoda, M. Ikeda, M. Hattori and J. Ichikawa, Synlett, 2013, 24, 57 CrossRef CAS PubMed; (d) J. Ichikawa, Y. Wada, M. Fujiwara and K. Sakoda, Synthesis, 2002, 13, 1917 CrossRef PubMed; (e) K. Fuchibe, M. Takahashi and J. Ichikawa, Angew. Chem., Int. Ed., 2012, 51, 12059 CrossRef CAS PubMed; (f) J. A. Cooper, C. M. Olivares and G. Sandford, J. Org. Chem., 2001, 66, 4887 CrossRef CAS PubMed; (g) F. Tellier, M. Audouin and R. Sauvêtre, J. Fluorine Chem., 2002, 113, 167 CrossRef CAS.
- (a) J. D. Moody, D. VanDerveer, D. W. Smith Jr and S. T. Iacono, Org. Biomol. Chem., 2011, 9, 4842 RSC; (b) H.-K. Wu, J.-Q. Zhong, H.-M. Shen and H.-X. Shi, J. Fluorine Chem., 2013, 156, 5 CrossRef CAS PubMed; (c) A. E. Feiring, S. Rozen and E. R. Wonchoba, J. Fluorine Chem., 1998, 89, 31 CrossRef CAS; (d) B. T. Kim, Y. K. Min, N. K. Park, K. Y. Cho and I. H. Jeong, Heterocycles, 1995, 41, 641 CrossRef CAS.
- (a) A. E. Feiring and E. R. Wonchoba, J. Org. Chem., 1992, 57, 7014 CrossRef CAS; (b) A. Fuss and V. Koch, Synthesis, 1990, 7, 604 CrossRef; (c) M. M. Kremlev, A. I. Mushta and L. I. Moklyarchuk, Russ. J. Org. Chem., 2003, 39, 1125 CrossRef CAS; (d) E. Yang, M. R. Reese and J. M. Humphrey, Org. Lett., 2012, 14, 3944 CrossRef CAS PubMed; (e) J. Ichikawa, M. Kobayashi, N. Yokota, Y. Noda and T. Minami, Tetrahedron, 1994, 50, 11637 CrossRef CAS; (f) R. Arnold-Stanton and D. M. Lemal, J. Org. Chem., 1991, 56, 151 CrossRef CAS.
- (a) Y. Xiong, X.-X. Zhang, T. Huang and S. Cao, J. Org. Chem., 2014, 79, 6395 CrossRef CAS PubMed; (b) W.-P. Dai, J. Xiao, G.-Y. Jin, J.-J. Wu and S. Cao, J. Org. Chem., 2014, 79, 10537 CrossRef CAS PubMed; (c) G.-Y. Jin, J. Zhang, W. Wu and S. Cao, J. Fluorine Chem., 2014, 168, 240 CrossRef CAS PubMed; (d) X.-X. Zhang, Y.-Y. Lin, J. Zhang and S. Cao, RSC Adv., 2015, 5, 7905 RSC.
- (a) D.-S. Chen and J.-M. Huang, Synlett, 2013, 24, 499 CrossRef CAS PubMed; (b) S.-M. Guo, L. Lu and H. Cai, Synlett, 2014, 24, 1712 CrossRef PubMed; (c) Y.-Q. Zou, J.-R. Chen, X.-P. Liu, L.-Q. Lu, R. L. Davis, K. A. Jørgensen and W.-J. Xiao, Angew. Chem., Int. Ed., 2012, 51, 784 CrossRef CAS PubMed; (d) C. Zhu, R. Wang and J. R. Falck, Org. Lett., 2012, 14, 3494 CrossRef CAS PubMed.
- (a) M. Moreno-Mañas, M. Pérez and R. Pleixats, J. Org. Chem., 1996, 61, 2346 CrossRef; (b) J.-Q. Liu and M. J. Robins, Org. Lett., 2004, 6, 3421 CrossRef CAS PubMed; (c) M. A. Aramendía and F. Lafont, J. Org. Chem., 1999, 64, 3592 CrossRef PubMed.
- J. Zhang, J.-J. Wu, Y. Xiong and S. Cao, Chem. Commun., 2012, 48, 8553 RSC.
Footnote |
| † Electronic supplementary information (ESI) available. See DOI: 10.1039/c4ra16468g |
| This journal is © The Royal Society of Chemistry 2015 |