
Polymer/gold hybrid nanoparticles: from synthesis to cancer theranostic applications
Xingjie Wu,
Yanqin Gao and
Chang-Ming Dong*
Department of Polymer Science & Engineering, School of Chemistry & Chemical Engineering, Shanghai Jiao Tong University, Shanghai 200240, P. R. China. E-mail: cmdong@sjtu.edu.cn
First published on 8th January 2015
Abstract
Gold nanoparticles (AuNP) with excellent optical and localized surface plasmon resonance properties have received intensive attention in biomedical applications. To achieve good therapeutic efficacy and minimize side effects in cancer treatment, increasing efforts have been made to combine therapeutic, imaging and targeting functions together in one drug delivery system. Polymer/gold hybrid nanoparticles are appealing for cancer treatment because they can be used as various theranostic modalities, including photothermal therapy (PTT), photodynamic therapy (PDT), chemotherapy, and imaging probes as well. Furthermore, the combination of more than one therapy methods that shows synergistic effect and good therapeutic efficacy in cancer treatment can be achieved by the polymer/AuNP hybrids. This minireview highlights the preparation methods for various polymer/AuNP hybrids and the recent progress on their cancer theranostic applications.
1. Introduction
In the 21st' century, cancer is becoming the biggest threat to human health. Surgical operations and traditional cancer therapies including radiotherapy, chemotherapy and hyperthermia have been widely used to treat patients. However, the traditional methods lack spatiotemporal control over treatment, which often causes serious damage to healthy tissues while therapeutic dosage reaches the optimum level. Thus, the development of novel cancer therapy strategy with lower cost, less side effect and optimal therapeutic efficacy is demanding. To realize the spatiotemporal control over cancer treatment and achieve the synergistic effect of more than one therapy methods are demonstrated to be promising tools for new generation cancer therapy.1Owing to the localized surface plasmon resonance (LSPR), AuNP can absorb and scatter incident light at specific wavelength. By varying the shape, size and morphology, the LSPR spectrum of AuNP can be tuned into the near infrared (NIR, 650–1000 nm) light region.1,2 Owing to its lower absorbance and scattering properties, the NIR light can penetrate deeply into the human tissues, enabling it promising as a noninvasive and harmless tool for clinical applications. AuNPs, such as gold nanoshells (AuNS), nanorods (AuNR) and nanocages (AuNC) have demonstrated to be excellent photothermal therapy (PTT) modalities because of their strong NIR adsorption and high photothermal conversion efficiency. AuNPs are also ideal radiosensitizers, which can generate secondary electrons under X- or γ-ray radiation leading to DNA and protein breakdown.1–4 On the other hand, AuNPs are applicable for various imaging techniques, including dark fields microscopy (DFM), optical coherence tomography (OCT), computed tomography (CT), and photoacoustic tomography (PAT) as well.1,5 These attractive properties make AuNP ideal platform for developing the theranostic modalities combining therapeutic, targeting and imaging functions while possessing synergistic effect of multi-therapies.
Biopolymers are widely used to improve the biocompatibilities of metal NPs and the polymer/AuNP hybrids are more stable and less toxic to human body. Furthermore, stimuli-responsive polymers render AuNP sensitive to stimulus, such as pH, temperature, redox or light. Moreover, functional biopolymers can further endow AuNP with specific binding properties, and/or imaging properties such as magnetic resonance imaging or ultrasound imaging.5 Based on this philosophy, various polymer/Au hybrid systems have been developed recently. For example, various biocompatible polymers, including poly(lactic-co-glycolic acid) (PLGA), poly(ethylene glycol) (PEG) and polycaprolactone (PCL) were hybridized with AuNP to generate the related polymer/AuNP hybrids.5 In this minireview, the preparation methods for polymer/AuNP hybrids, including surface coating, AuNP self-assembly and gold deposition are first discussed. In the following section, we briefly give an overview on the therapeutic and imaging applications of these polymer/AuNP hybrids.
2. Preparation of polymer/gold hybrid nanoparticles
2.1. AuNP surface coating
In most cases, the as-synthesized AuNPs are capped by surfactants such as hexadecyltrimethylammonium bromide (CTAB) to prohibit severe aggregation in aqueous solution. However the surfactants stabilizing AuNPs are often found to be toxic in vivo. It is necessary to cap AuNP with other biomolecules or biocompatible polymers, which is generally achieved by layer-by-layer electrostatic self-assembly or ligand-exchange reaction with surfactants. The thiol–gold (S–Au) coordination interactions are usually used to replace various polymers with CTAB.6–9 By esterification with the thiol-containing molecules, various kinds of biocompatible polymers can be readily coated onto AuNP surfaces. Biodegradable diblock copolymer PEG-b-PCL terminated with one disulfide group was added dropwise into AuNR suspension and then was successfully coated on AuNR surfaces after stirring for 4 h.6 Similarly, 3-mercaptopropionic acid modified dextran was used for the preparation of the dextran coated AuNR (DEX-AuNR), exhibiting 1-fold higher cell uptake efficiency and similar toxicity on RAW 264.7 cells compared to PEGylated AuNR (Fig. 1).7 In comparison to PEGylated AuNR, DEX-AuNR revealed excellent photothermal ablation efficacy at a lower dose and laser power, demonstrating potential for the treatment of inflammatory macrophages related to diseases. Obviously, more work is needed to be carried out for further clinical applications. Note that the coating of dextran to some extent attenuates the photothermal property of AuNR. Similar result was obtained when AuNR was wrapped with polyelectrolytes through electrostatic interactions. The adsorption intensity at 800 nm dropped evidently as more polyelectrolytes layers were adsorbed onto AuNR surfaces.10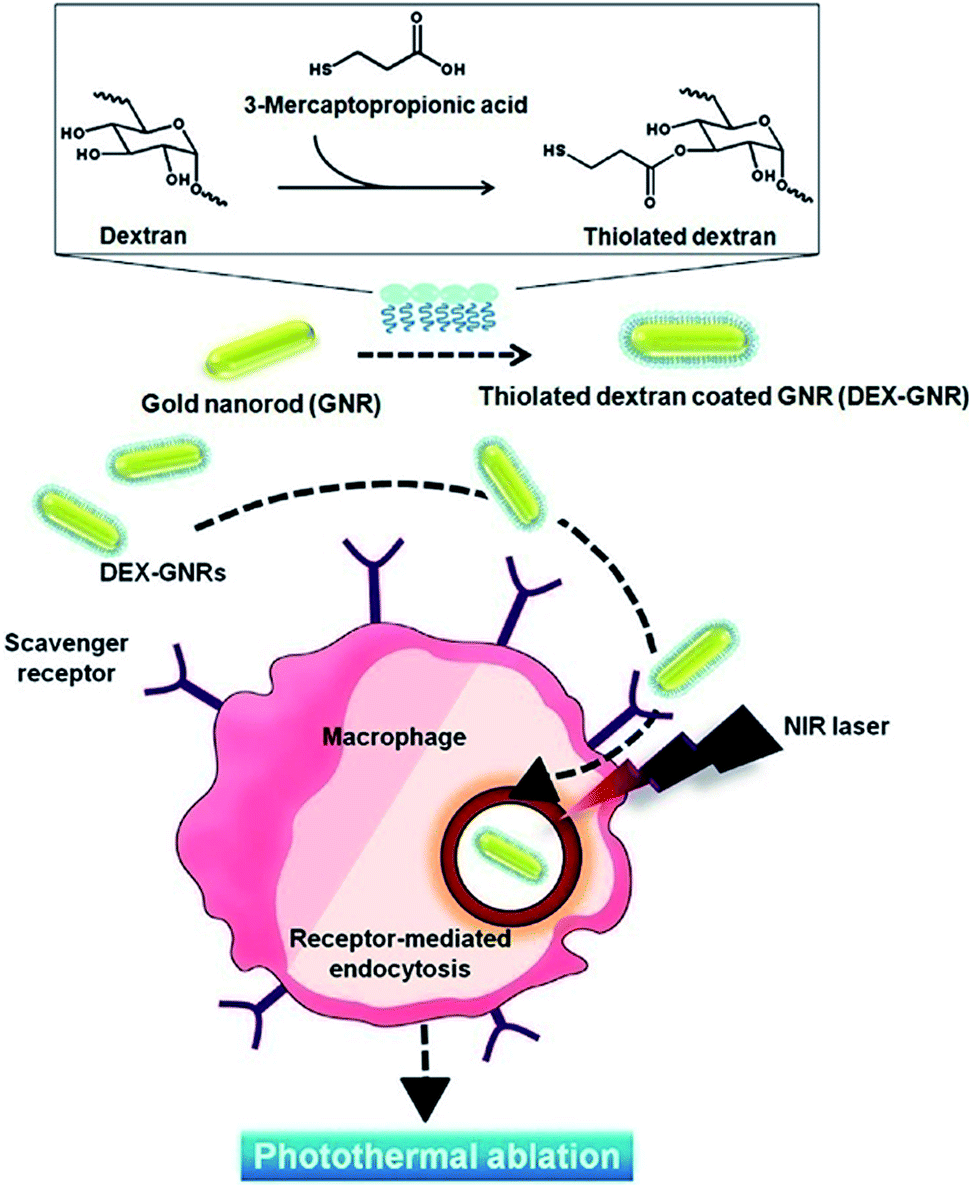 | ||
| Fig. 1 Schematic illustration of thiolated dextran-coated gold nanorods (DEX-GNR) synthesis. Adapted from ref. 7. | ||
Surfactants can also be replaced by the thiol-modified initiators used for atom transfer radical polymerization (ATRP), which can initiate polymerization directly on AuNP surfaces.11–15 Chen et al. used N-isopropylacrylamide (NIPAAm), acrylic acid and N,N′-methylenebis(acrylamide) (MBA) monomers together to polymerize thermo- and pH-responsive units on SiO2 coated AuNR (Au@SiO2) and successfully obtained temperature- and pH-responsive AuNR.14 The blood circulation time of poly(NIPAAm-c-acrylic acid-c-MBA) modified AuNR was dramatically prolonged compared with bovine serum albumin (BSA) coated AuNR and Au@SiO2. Pyrrole (Py) can be polymerized to produce polypyrrole (PPy) under the oxidation of (NH4)2S2O8, which is known to be a conductive polymer and is applied as photothermal agent. By vigorously stirring Py, (NH4)2S2O8 and urchinlike AuNP at room temperature, the PPy coated urchinlike AuNP hybrid can be obtained conveniently (Fig. 2).15 Compared with bare AuNP, PPy-coated AuNP exhibited improved structural stability toward storage, heat, pH, and laser irradiation. Further investigations revealed that the thin shell of PPy can enhance the photothermal conversion efficiency (η) of PPy-coated AuNP, resulting from the absorption of PPy in the red and NIR light regions. Specifically, the PPy-coated AuNP with a Au core diameter of 120 nm and a PPy shell of 6 nm exhibited a higher η of 24.0% at 808 nm than that (η = 11.0%) of bare AuNP.
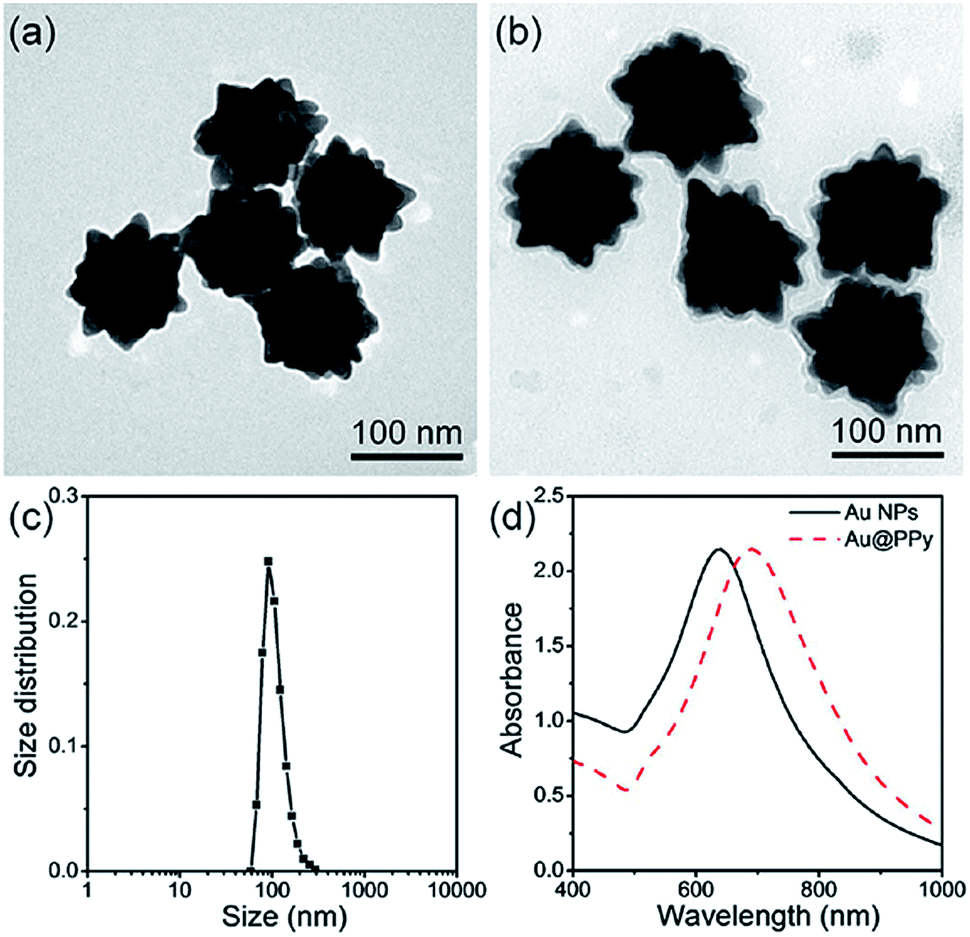 | ||
| Fig. 2 TEM images of the urchinlike Au NP (a) before and (b) after the PPy coating. (c) DLS size distribution of PPy-coated urchinlike Au NP. (d) UV-vis spectra of the urchinlike Au NP before and after the PPy coating. Adapted from ref. 15. | ||
2.2. AuNP self-assembly
AuNP self-assemblies are fascinating due to their enhanced electromagnetic properties and collective effects compared with the bulk materials. AuNP can act as a hydrophobic block of amphiphilic copolymers and provide a promising tool for the fabrication of various AuNP assemblies. AuNP with a diameter of 2–4 nm was anchored with one triblock copolymer consisting of PEG and polystyrene (PS) as outer blocks and poly(lipoic acid-2-hydroxy-3-methacryloyloxy-propylester-co-glycidyl methacrylate) [P(LAMP-co-GMA)] as inner block.16 Such anchoring is demonstrated to be stoichiometric that one polymer chain was wrapped around one AuNP. Similar to amphiphilic copolymers, PEG–AuNP–PS formed a variety of assemblies in water. By varying PEG/PS ratio, different morphologies, including hybrid micelles, (branching) rods, vesicles, and large compound micelles can be obtained (Fig. 3). The interparticle coupling of self-assembled AuNP usually leads to collective properties, which might improve LSPR adsorption in NIR light region and photothermal properties. Note that the photothermal properties of these hybrid assemblies were not further investigated. In another report, the ligand exchange technique and surface ATRP method were applied in a stepwise manner to coat PEG and poly(methyl methacrylate) (PMMA) on AuNP surface, respectively.17 The as-synthesized copolymer brush coated AuNR assembled into vesicles in water and possessed LSPR peak in NIR light region. Under the NIR light irradiation for 2 min (785 nm laser at 2 W cm−2), the PMMA shells collapsed as a result of the generated heat, and the assembled AuNR changed from spherical morphology to flattened aggregates. The same strategy was also applied to construct anticancer drug delivery systems and multi-modal imaging contrast agents.18,19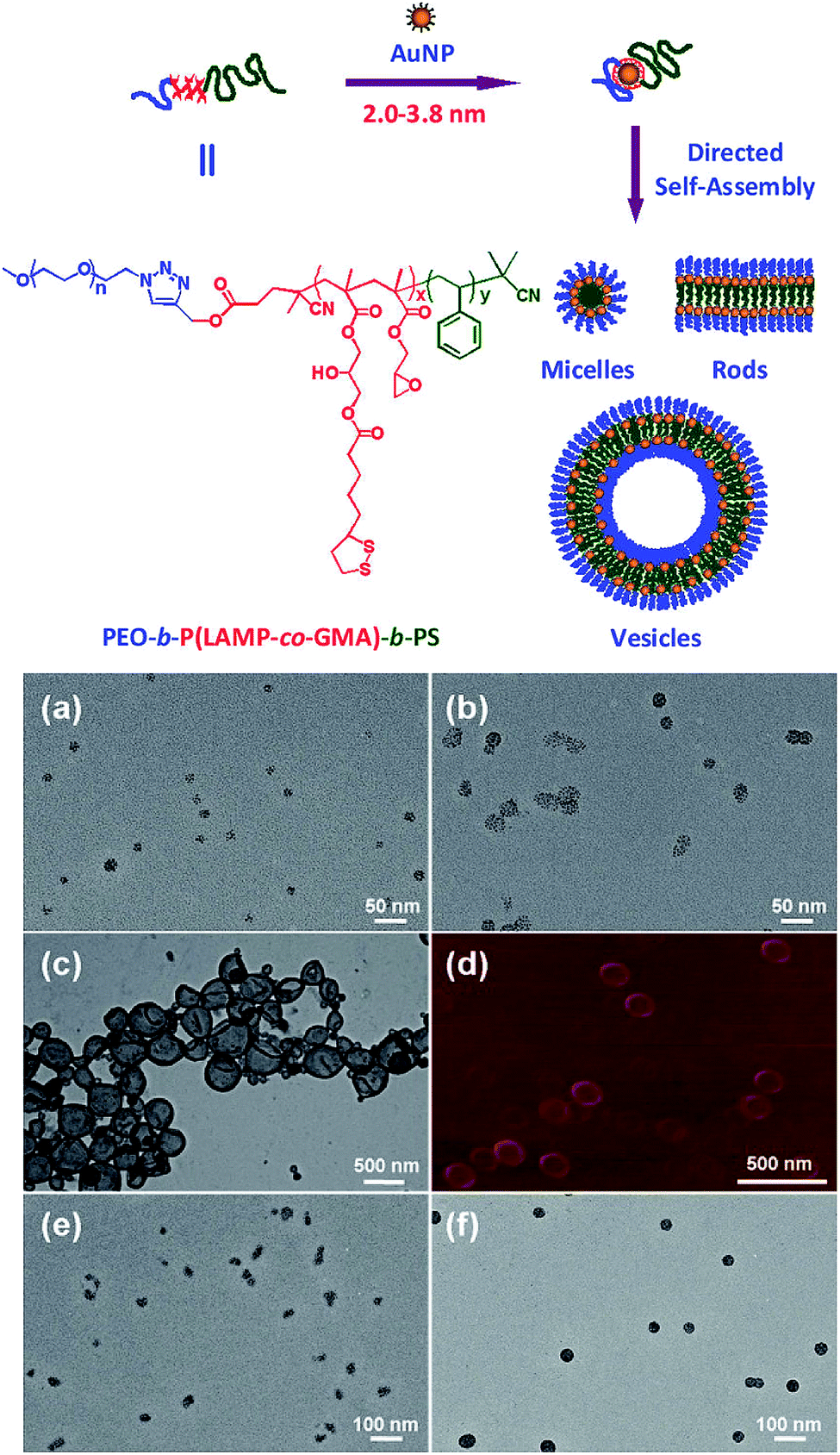 | ||
| Fig. 3 Fabrication of ultrasmall AuNPs anchored with a single triblock copolymer and the hierarchical self-assembly of the AuNP hybrid amphiphilic triblock copolymer. (a–c, e and f) HRTEM images and (d) AFM height imagerecorded for hierarchical NP assemblies fabricated from AuNP (2.0 ± 0.2 nm) hybrid amphiphilic triblock copolymers in aqueous solution: (a) PEO45–AuNP2.0–PS45; (b) PEO45–AuNP2.0–PS135; (c and d) PEO45–AuNP2.0–PS260; (e) PEO113–AuNP2.0–PS60; (f) PEO113–AuNP2.0–PS300. Adapted from ref. 16. | ||
In addition, AuNP self-assembly can also be controlled by pH20,21 or temperature.22–24 In a recent work, spherical AuNP (14 nm in diameter) was “polymerized” to generate AuNP chain by simply mixing pyrrole with AuNP.25 The polymerization was terminated by the addition of (NH4)2S2O8 and PPy layer was formed around AuNP chain. By adjusting the pyrrole content or polymerization time, the AuNP chain length could be tuned. The photothermal conversion efficiency of AuNP chain was enhanced to 70% as AuNP chain length grew up to 10 AuNP.
2.3. Gold deposition
Gold can also be deposited on polymer NP surface to form AuNS. Traditional AuNS that formed on silica core has been extensively studied and is well known for its outstanding photothermal properties. However, silica cannot degrade in vivo, which limits its further clinical applications to some extent. To overcome this obstacle, biodegradable polymers such as PLGA and polylactide were used to replace silica.26,27 Similar to the preparation of AuNS on silica core, small gold nanoclusters are attached to polymer NP surface and can act as nucleation site for further reduction of Au ions into Au(0) by reductants, such as formaldehyde, sodium borohydride and sodium citrate tribasic. Thus, thick gold shells that lead to higher photothermal conversion efficiency can be deposited on polymer NP surface. Cysteamine modified doxorubicin (DOX)-loaded PLGA NP was deposited with 3 nm AuNP and was further coated with a gold nanolayer by reduction (Fig. 4).26 An anti-EGFR antibody Cetuximab (CET) was then decorated on AuNS surface as a targeting moiety and signal transduction inhibitor. After being treated with CET modified AuNS, A431 cells (EGFR-abundant) showed six times higher fluorescence intensity than MCF7 cells (EGFR-deficient). The difference of fluorescence intensity between the two cell lines was in accordance with that in EGFR expression level. Dong et al. utilized biodegradable block copolymer PCL-b-poly(L-lysine) self-assembled spherical micelles as template, and HAuCl4 was reduced on the surfaces of these micelles by formaldehyde.28 It was demonstrated that a thin and smooth gold shell formed on the surface of the micelles at pH >10, at which the deprotonated primary amines of poly(L-lysine) block were coordinated with Au(0).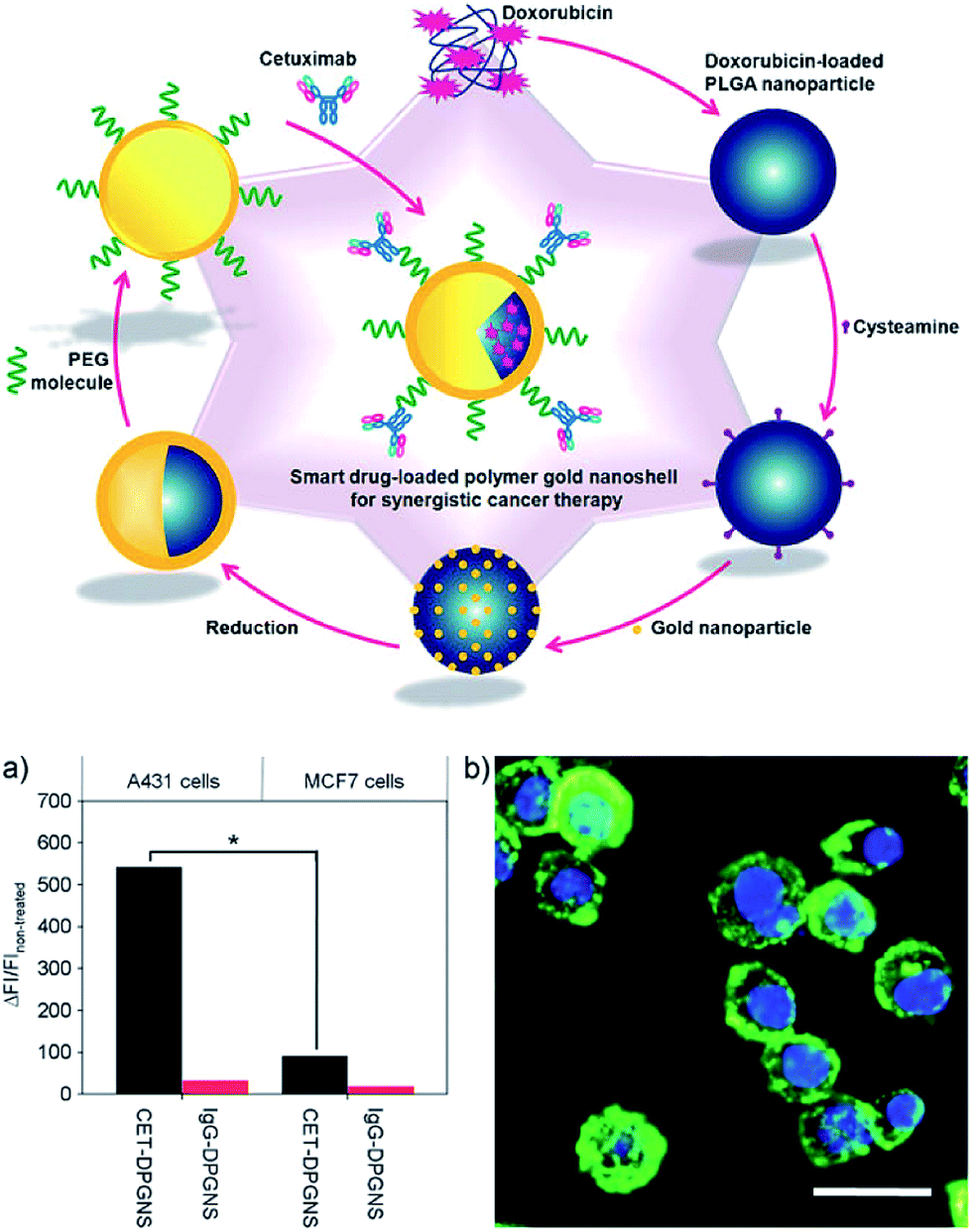 | ||
| Fig. 4 Schematic illustration of multifunctional drug-loaded gold nanoshells for synergistic cancer therapy. The novel nanostructure comprises three key parts for multidimensional therapeutic potentials, namely, (I) an anti-EGFR antibody, Cetuximab (CET), as a targeting moiety and signaltransduction inhibitor, (II) a gold nanoshell that has a photothermal effect due to plasmon resonance upon illumination by an NIR laser, and (III) DOX as a chemotherapeutic agent. These three components, incorporated into PLGA nanoparticles, provide synergistic tumoricidal efficacy. (a) Relative fluorescence intensities (DFI/FInon-treated; FI: fluorescence intensity) using flow cytometry analysis (*p < 0.05). (b) Fluorescence microscopic images of A431 cells treated with CET-DPGNS. The bright green-light emission from binding of fluorescein isothiocyanatelabeled goat anti-CET antibody indicates successful binding of CET-DPGNS to target cells, and the blue indicates cell nuclei. Scale bar: 20 mm. Adapted from ref. 26. | ||
Electron beam evaporation was applied to directly deposit gold on NP surface, which enabled researchers to precisely control the thickness of AuNP.29 Different thickness of Au half-shells (15–25 nm) were deposited onto monolayers of NPs casted on a silicon substrate by the evaporation method (Fig. 5). The gold layers outside the AuNS surfaces contributed to the photothermal abilities. However, in most cases, hydrophobic drug inside AuNS often diffuses through gold shells at a negligible rate, which dwarfs the synergistic combination strategy of chemo-photothermal therapy. The half shell structure is a smart design to overcome this drawback. To combine the chemotherapy mediated by the anticancer drug DOX and PTT, DOX-loaded poly(lactic-co-glycolic acid)–gold half-shell nanoparticles (DOX-loaded PLGA–Au H-S NP) were fabricated by depositing Au films on DOX-loaded PLGA NP. DOX was released with the biodegradation of PLGA core and heat was generated upon NIR light irradiation. Compared with chemotherapy or PTT alone, the combined treatment that was achieved by DOX-loaded PLGA–Au H-S NP demonstrated a synergistic effect, resulting in higher therapeutic efficacy and shorter treatment time.29 Studies on animal further showed that the combined DOX and PTT treatment resulted in complete destruction of tumors without weight loss or recurrence of tumors, and the chemo-photothermal treatments based on DOX-loaded PEG–PLGA–Au H-S NP were superior to chemotherapy or PTT alone.30 Similarly, methotrexate (MTX)-loaded PLGA Au half-shell nanoparticles (MTX–PLGA–Au) decorated with arginine-glycineaspartic acid (RGD) peptides on the surface of Au half-shell were developed for the treatment of rheumatoid arthritis (RA), which also has potential application for other inflammatory diseases.31
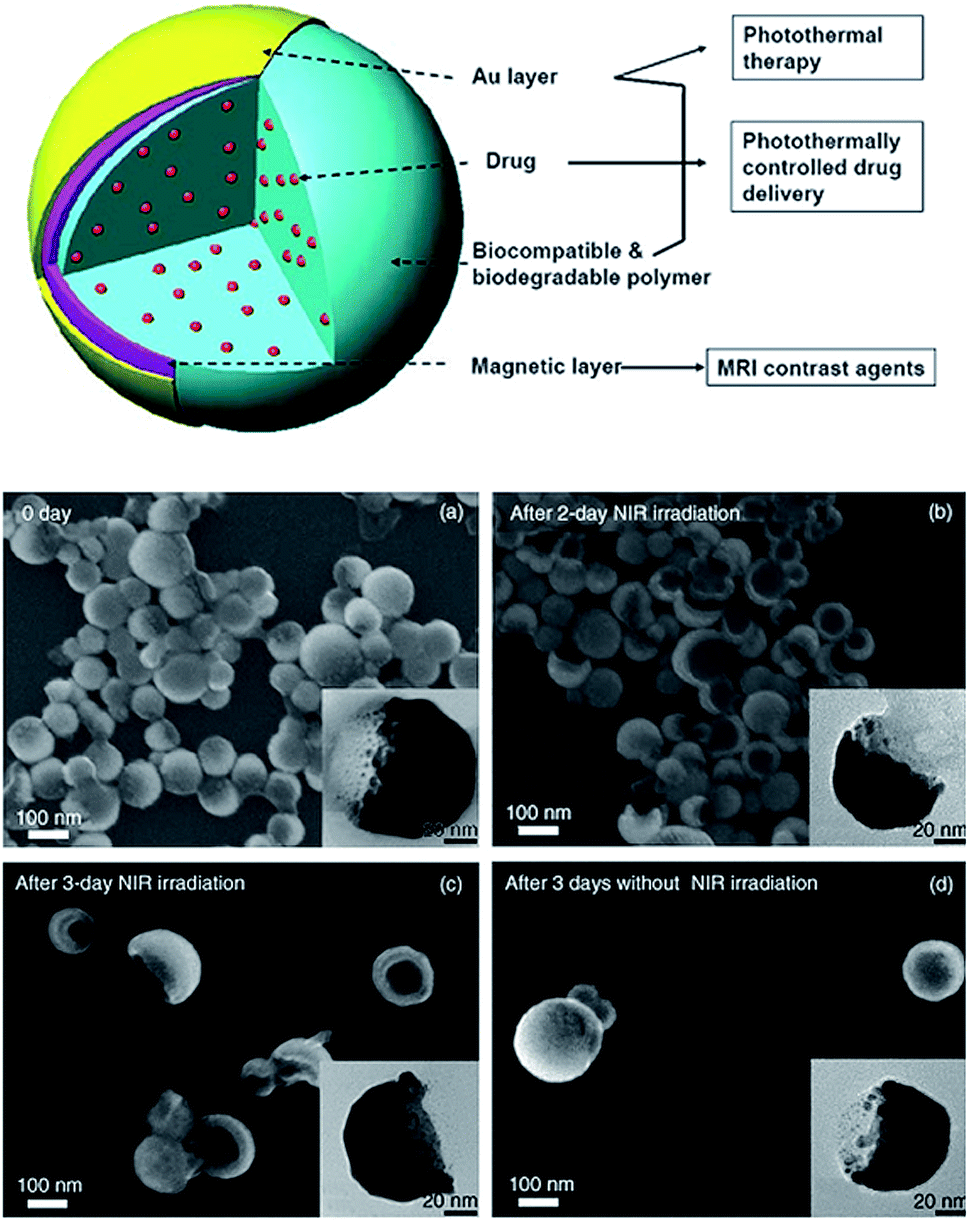 | ||
| Fig. 5 Schematic diagram of drug-loaded polymer–metal multilayer H-S NP. The drug is loaded into biocompatible and biodegradable polymer nanoparticles, and magnetic and Au layers are deposited on the polymer nanoparticles. These nanoparticles provide multiple functions, such as photothermal therapy, photothermally controlled drug delivery, and MRI contrast enhancement. FESEM images of PLGA–Au H–S NP right after fabrication (a), after 2 day NIR irradiation (b), after 3 day NIR irradiation (c), and in water for three days without NIR irradiation (d). The insets areTEM images. Adapted from ref. 29. | ||
3. Cancer theranostic applications
Taking the advantages of their unique optical properties, the polymer/AuNP hybrids have aroused tremendous interest as therapeutic and multi-modal imaging agents especially for cancer therapy (Scheme 1). For example, the polymer/AuNP hybrids can absorb and dissipate light energy thermally to induce tumor tissue hyperthermia, such procedure also known as PTT, is emerging as a noninvasive and powerful method for photothermal cancer therapy due to its enhanced spatiotemporal control and lower side effects in the diseased sites.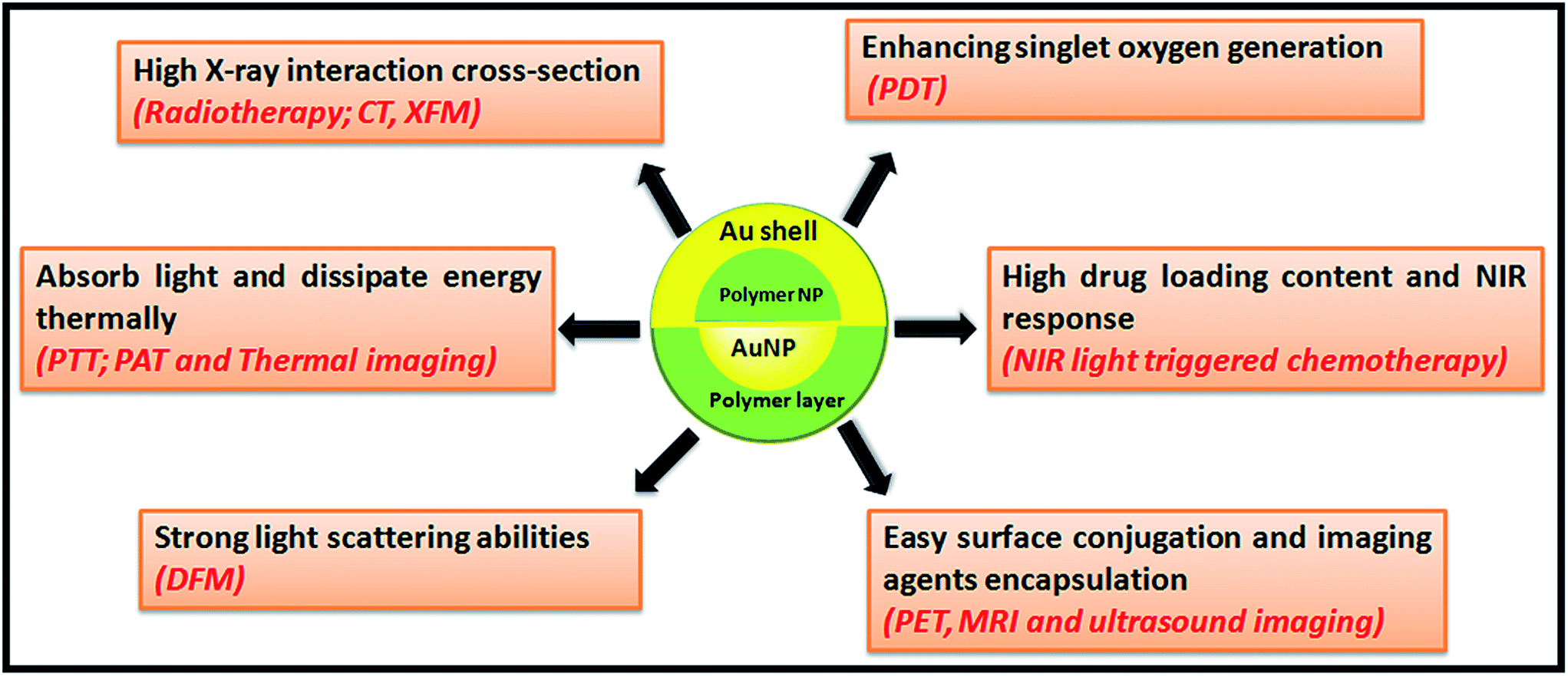 | ||
| Scheme 1 Illustration for various biomedical applications of the polymer/AuNP hybrids. | ||
Due to AuNP's large surface area, anticancer or image contrast agents can be easily conjugated or physically adsorbed onto polymer/AuNP hybrids, which enable the synergistic combination of different therapy methods and multi-modal imaging practicable. Herein, we will briefly discuss about the therapeutic and multi-modal imaging properties of polymer/AuNP hybrids.
3.1. Cancer therapies
Anticancer drugs can be entrapped inside surfactant layers surrounding AuNP or encapsulated in polymer brushes on AuNP surface or covalently anchored to AuNP. When drugs are entrapped in polymer/AuNP hybrids possessing photothermal properties, the combined chemotherapy and PTT can be achieved and the drug release profile can also be adjusted by NIR light irradiation.6,32–36 The heat generated by NIR light irradiation can accelerate drug release by enhancing polymer degradation or drug diffusion. For instance, DOX, as a first-line clinical anticancer drug, is loaded in PEG-b-PCL coated AuNR.6 The DOX release can be accelerated from 7% to 68.3% by NIR light irradiation (808 nm, 0.2 W cm−2) for 5 min at 1, 2, 3 and 4 h, respectively and an NIR-triggered “on–off” release mode was demonstrated. Furthermore, if the sensitive polymers with the volume phase transition properties in physiological environments are used to prepare NP, a smart load-and-release profile can be achieved. Thermosensitive poly(NIPAAm-co-acrylamide) brushes were coated on the surface of gold nanocage by ATRP method and alizarin-PEG was encapsulated in AuNC.32 After exposing to NIR laser at a power density of 10 mW cm−2 for 16 min, most of the entrapped alizarin-PEG was released. The AuNC was able to reload alizarin-PEG and the same release amount as the first round release profile was obtained. In another report, poly(NIPAAm-co-acrylamide) covered AuNC was prepared by the ligand exchange between poly(NIPAAm-co-acrylamide) and PVP, and fluorescence, prodrugs, enzymes or inhibitors were loaded within AuNC separately (Fig. 6).33 As the AuNC LSPR peaks can be adjusted by changing Au/Ag alloy aspect ratio, the orthogonal release of the loaded contents and the remotely logic activation of prodrugs could be realized by NIR light irradiation.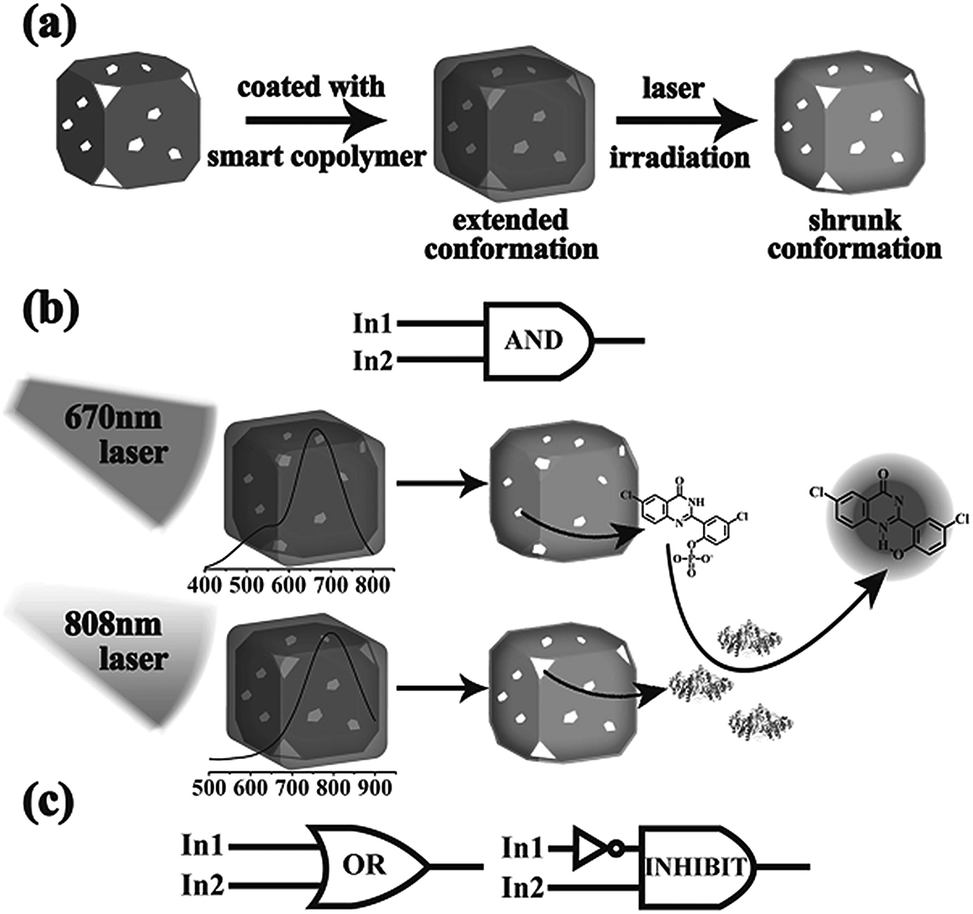 | ||
| Fig. 6 (a) Illustration of photothermal-sensitive AuNC-copolymer. (b and c) Schematic representation of a NIR light-encoded logic gate for controlled release based on AuNC-copolymer. Adapted from ref. 33. | ||
PDT requires the illumination of photosensitizer to generate reactive singlet oxygen, which is deadly poisonous to cells. The ability to generate singlet oxygen by photosensitizer can be enhanced by electromagnetic waves of irradiating AuNP.37–42 A phthalocyanine-based photodynamic prodrug Pc 227 was conjugated to PEG decorated AuNP surface through thiol–Au interactions.38 Upon irradiation with Vis-NIR light, Pc 227 was cleaved from AuNP surface and activated to photosensitizer drug Pc 4. Upon irradiation at 660 nm and 1 J cm−2, AuNP conjugated 227 showed evident higher depression effect against HeLa cells than free Pc 227. In another report, aptamer switch probe (ASP) linking chlorine e6 (Ce6) was conjugated to AuNR.39 ASP structure was switchable and could activate Ce6 in the presence of targeted cells, whereas Ce6 was quenched on the surface of the AuNR (Fig. 7). In this way, the unexpected release of photosensitizer in normal cells and photodynamic damage to healthy tissues would be minimized. This PDT/PTT multimodal strategy proved to be an efficient therapeutic regimen against cancer cells than single PDT or PTT because of high selectivity and specificity of ASP.
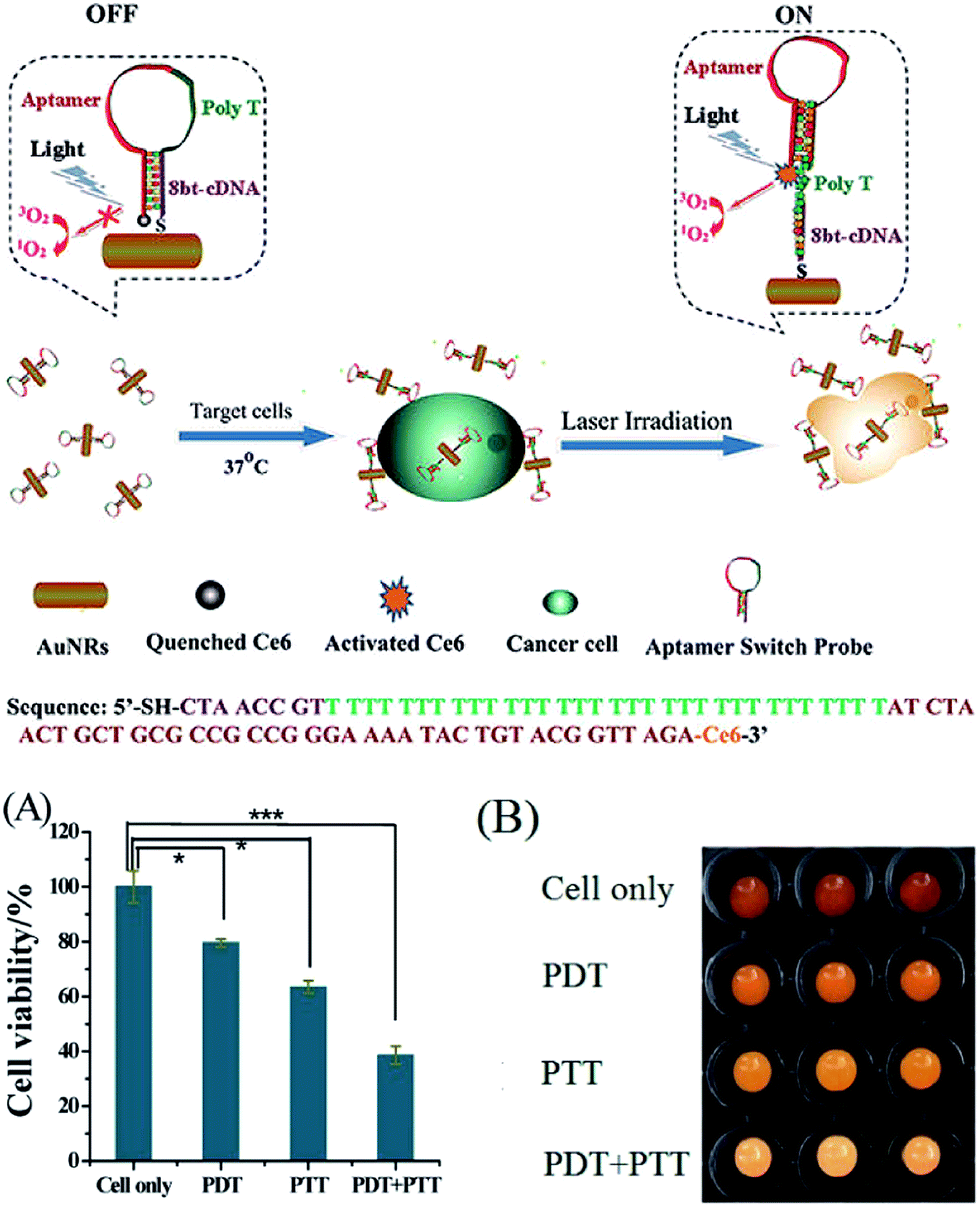 | ||
| Fig. 7 Schematic representation of ASP-photosensitizer-AuNRs for PTT and PDT. Cell viability data (A) and imaging (B) of CCRF-CEM cells incubated with Ce6-ASP-T32-NR without light irradiation (cells only), under white light irradiation (PDT), under 812 nm laser irradiation (PTT), and PDT + PTT, respectively. Adapted from ref. 39. | ||
When exposing to X- or γ-ray radiation, human tissues generate secondary electrons, which can break down proteins and DNA in tumor. To enhance therapeutic index of radiotherapy, radiosensitizers are used. Gold is known to have high atomic number, which leads to higher X-ray interaction cross-section than elements with low atomic number and AuNPs are proved to be attractive radiosensitizers.43–45 For example, 1.9 nm AuNP was loaded into PEG-b-PCL micelles to form gold polymeric micelles (GPMs) and tested as radiosensitizer in vivo.43 It was demonstrated that GPMs could remarkably enhance radiation-induced DNA breakdown, improve the accumulation in tumor xenografts, and induce a clear CT contrast. Consequently, the combination of CT-guided radiation therapy and gold-mediated radiosensitivity significantly increased the mean survival time of tumor-bearing mice.
AuNPs are also developed to deliver nucleic acids for controlling the tumor-related genetic response or protein expressions.46–49 Ethyleneglycoldiglycidylether (EGDE) cationic polymer or poly(ethyleneimine) (PEI) was electrostatically deposited onto poly(styrene sulfonate) (PSS) coated AuNR.46 Both kinds of polyelectrolytes coated AuNR showed excellent stabilities even after 4 weeks of storage. pGL3 plasmid DNA was loaded within these positively charged AuNRs and EGDE-polymer coated AuNRs were demonstrated to have lower cytotoxicities and higher transfection efficacies compared with PEI coated AuNR. Halas et al. modified one strand of a double-stranded DNA (dsDNA) with thiol moieties, and conjugated dsDNA on AuNR or AuNS through gold–thiol interactions.47 Upon 800 nm laser illumination, the dsDNA on AuNS surface was dehybridized in a nonthermal manner as the solution temperature was far below DNA melting temperature; meanwhile, the nonthiolated DNA strand was released from AuNS surface. However, only thermal-induced release was observed for AuNR. The mechanism for light-induced manner involved the transfer of hot electrons from AuNS surface to dsDNA, which was able to induce DNA dehybridization. The differences in adsorption cross-section and dsDNA densities made such phenomenon only observable for AuNS. Short-interfering RNA (siRNA) or dsDNA were loaded within AuNS by hybridization with poly(L-lysine) by the same group. Then, the loaded genes were successfully transported to the targeted cells and released from AuNS surface by NIR light irradiation.48 Moreover, the down regulation of green fluorescent protein (GFP) in H1299 GFP/RFP cell line by antisense ssDNA or siRNA was also found to be significantly enhanced by NIR light irradiation (Fig. 8).
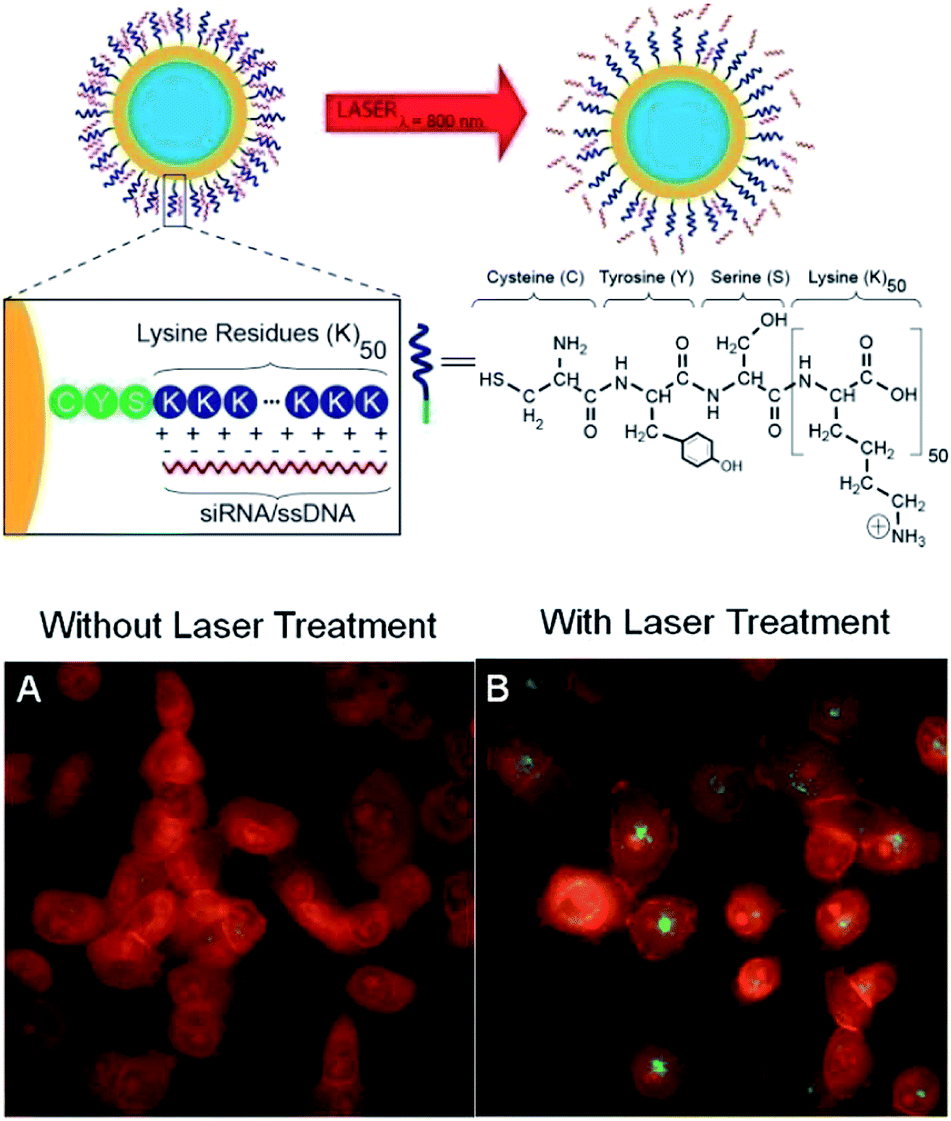 | ||
| Fig. 8 Au nanoshell poly-L-lysine (NS-PLL)-based therapeutic RNA delivery system. The negatively charged phosphate backbone of the siRNA/ssDNA (red) is electrostatically attached to the cationic peptide (blue), which consists of 1 cysteine, 1 tyrosine, 1 serine, and 50 lysine amino acids. Upon laser irradiation, the siRNA/ssDNA is released. Fluorescence images H1299 cells incubated with NS-PLL-ssDNA. The ssDNA is fluorescently tagged with Alexa Fluor 488 (green). (A) Without laser treatment, the green fluorescence is quenched because of the proximity of the ssDNA to the gold nanoshell surface. (B) With laser treatment, the ssDNA is released, which eliminates the quenching, resulting in brighter green fluorescence spread throughout the cell. Adapted from ref. 48. | ||
3.2. Imaging
AuNP proved to be excellent contrast agents for DFM and OCT because of their strong light scattering abilities. DFM is widely applied to image AuNP in vitro.48,50,51 Compared with traditional chromophore, the scattering properties of AuNP are far more stable, and AuNP with diameter more than 10 nm can be readily visualized by DFM.1 Halas et al. demonstrated whether polylysine modified gold nanoshells were endocytosed or remained outside H1299 cell by DFM and the number of nanoshells taken by per cell was determined by inductively coupled plasma mass spectrometry.48 In another work, streptavidin decorated AuNPs were bound to respiratory syncytial virus (RSV) surface through biotin–NHS interactions.50 The binding of AuNP to RSV surface did not affect its activities in Hep-2 cells. As AuNP–RSV hybrid was able to be visualized by DFM, the author successfully conducted the tracking of RSV infecting Hep-2 cells route in real-time (Fig. 9).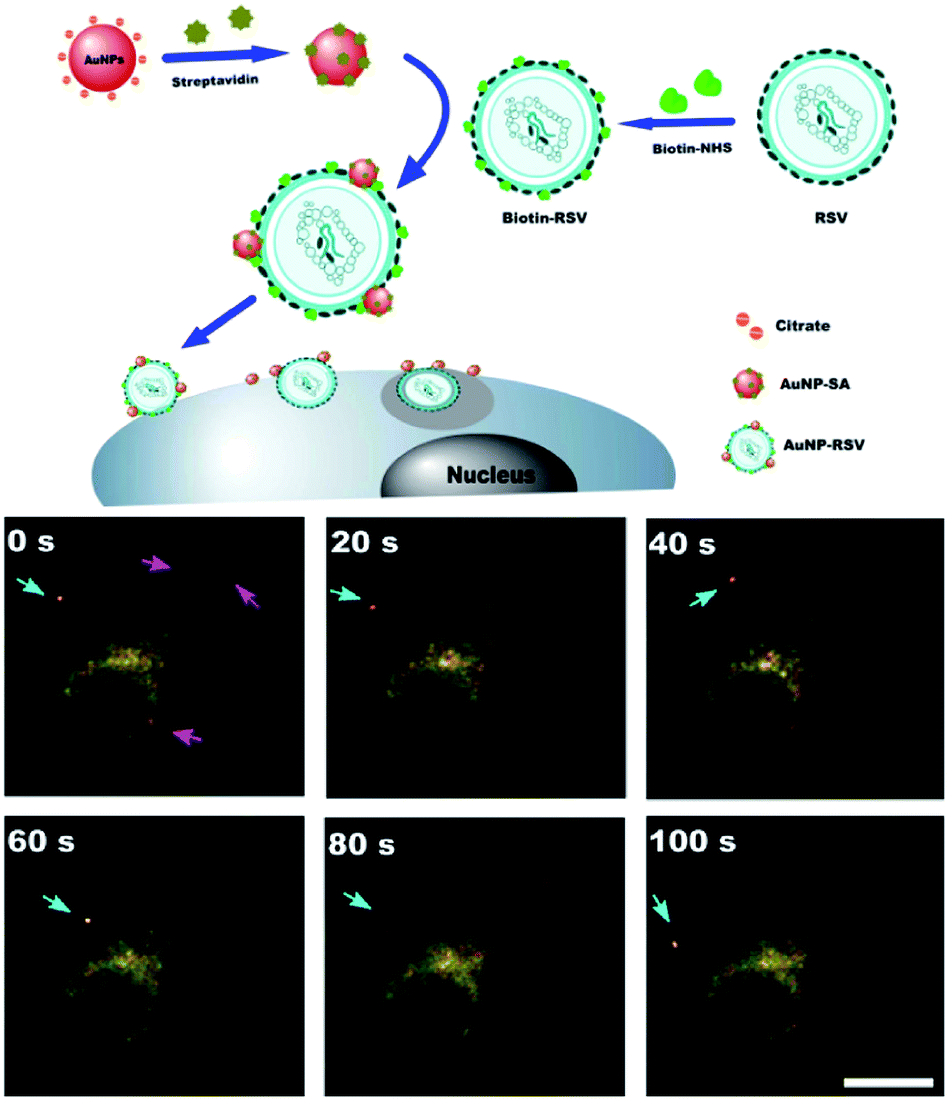 | ||
| Fig. 9 Schematic outlining the generation of AuNP conjugated with SA and RSV labeled with AuNP–SAs, then AuNP–RSVs invaded HEp-2 cells; real-time dark-field light scattering images of 13 nm AuNP–RSVs invading HEp-2 cells. The red arrow indicates a virus on the cell membrane when the 13 nm AuNP–RSVs are added to living HEp-2 cells. The blue arrow indicates the infection process for a single AuNP–RSV invading HEp-2 cells, which occurs over 1 min following the addition of AuNP–RSV. The scale bar indicates 20 mm. Adapted from ref. 50. | ||
Gold element with high X-ray interaction cross-section is used in X-ray related imaging techniques including CT52–54 and X-ray fluorescence microscopy (XFM).55 CT is a widely used imaging technique in the clinic and much effort has been made to develop contrast agents for improving CT resolution. Because of its larger surface area, smaller AuNP exhibited higher X-ray attenuation level. AuNP with 4 nm in diameter showed 25% higher X-ray attenuation than Omnipaque (a commercial iodinated-contrast agent) at Au concentration larger than 0.1 mol L−1, while X-ray attenuation was close to each other at low concentration (0.02 mg mL−1).52 In another work, PDMAEMA-grafted AuNR was successfully visualized by CT imaging in vitro, and the contrast of CT images was enhanced with increasing Au concentration.53 X-ray fluorescence is especially useful in the investigation of metals, glasses and ceramics. When coupled with synchrotron radiation technique, X-ray fluorescence mapping can be obtained. Thierry et al. used XFM and reflectance confocal microscopy to analyze the penetration depth of transferrin-conjugated AuNP in MCF-7 tumour spheroids.55 The results obtained by both techniques showed that the AuNP penetration was limited to 50 nm in 48 h.
AuNP can generate heat upon irradiation, which makes AuNP applicable in thermal imaging and PAT. As a frequently used method in the clinic, PAT is dependent on detecting acoustic waves produced by thermal expansion for its high contract and large penetration depth. Biodegradable gold vesicles (BGVs), which possessed a high photothermal conversion efficiency of 37% were fabricated from self assembly of PEG-b-PCL copolymer tethered AuNP through a dialysis method in water (Fig. 10).56 The in vivo photothermal therapy was monitored by PAT and thermal imaging. The temperature elevations of MDA-MB-435 tumor-bearing mice caused by BGVs irradiated by 808 nm laser at a power density of both 0.5 and 1 W cm−2 were recorded for different time intervals by thermal camera. The tumor temperature evidently increased over 18 °C in 5 min. After injection into tumor-bearing mice, BGVs showed approximately 10-fold stronger PA intensity than that was observed before injection. BGVs also had linear correlation between its concentration and PA signal intensity, which was much stronger than gold nanorods.
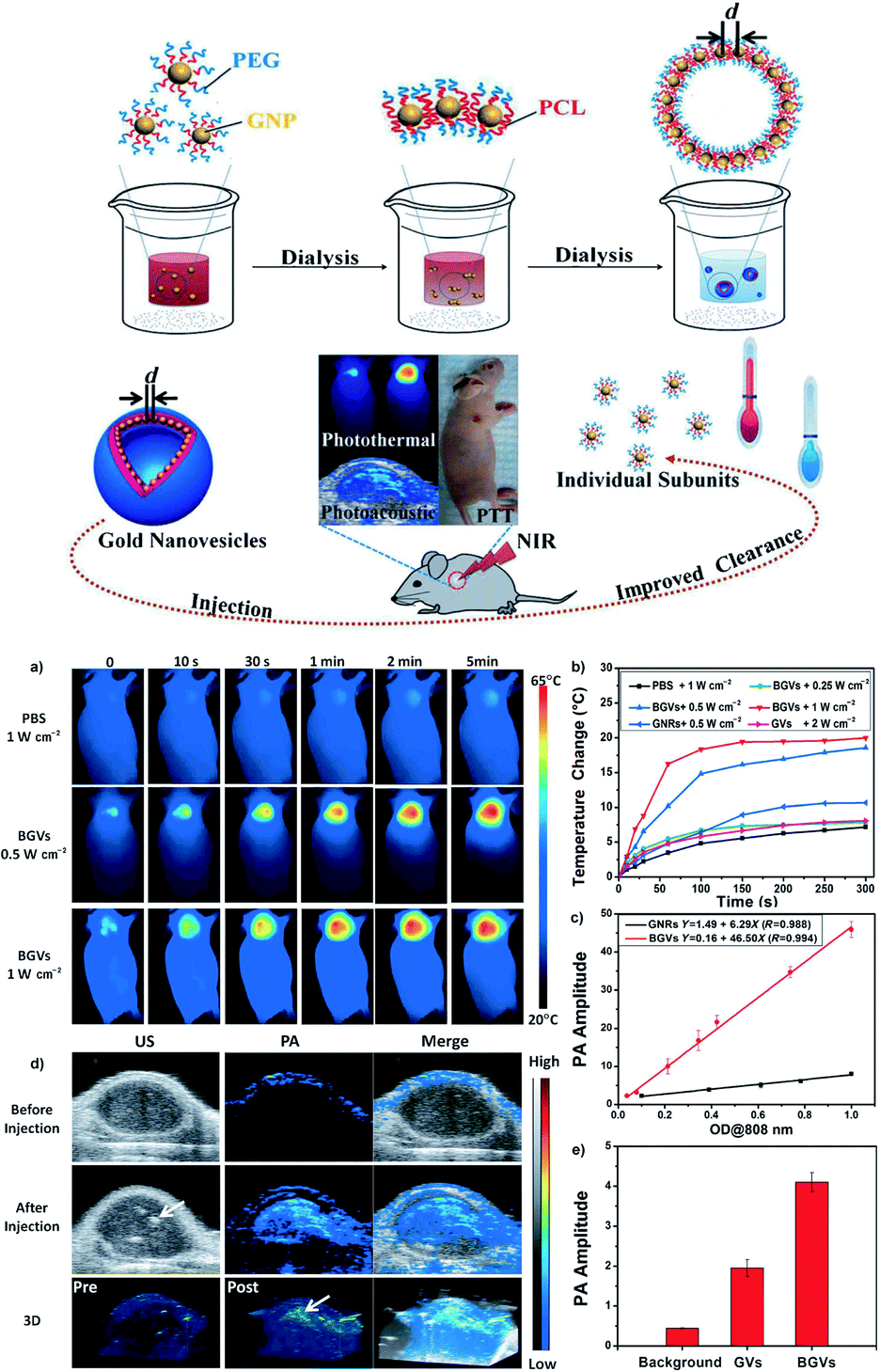 | ||
| Fig. 10 Self-assembly of biodegradable gold vesicles (BGVs) composed of PEG-b-PCL-tethered GNPs through the dot-line-plane-vesicle mode during the dialysis process. BGVs with an ultrastrong plasmonic coupling effect are superior PA imaging and PTT agents with improved clearance after the dissociation of the assemblies. (a) Thermal images of MDA-MB-435 tumor-bearing mice exposed to a 808 nm laser for 5 min after the injection of phosphate-buffered saline (PBS) or BGVs. (b) Heat curves of tumors upon laser irradiation as a function of irradiation time. (c) PA signals of BGVs and GNR as a function of optical density. (d) In vivo 2D ultrasonic (US) and photoacoustic (PA) images and 3D PA images of tumor tissues before and after the injection of BGVs. Arrows indicate the location of BGVs. (e) PA intensities of tumor tissues following the intratumoral administration of the same amount of GVs or BGVs. Adapted from ref. 56. | ||
Besides intrinsic imaging properties of AuNP, we can further conjugate AuNP with other contrast reagents to endow AuNP with new imaging functions or achieving multi-modal imaging. RGD peptide and 64Cu were simultaneously conjugated to PEG coated AuNR.57 The resultant AuNR could be visualized by positron emission tomography (PET) technique in U87MG tumor-bearing mice even after 45 h. The tumor tissue uptake ratio of RGD-modified AuNR was 8.37 ± 1.16% ID g−1 at 24 h post-injection, which was higher than 6.19 ± 0.5% ID g−1 for pure AuNR. MRI contrast agents, such as Fe3O4 and gadolinium were also entrapped within AuNP to realize MRI contrast. West et al. linked orthopyridyldisulfidepoly(ethylene glycol)-succinimidyl propionate (OPSS-PEG-SPA) with tetraazacyclododecanetetraacetic acid (DOTA), which is an aminated gadolinium chelator (Fig. 11).58 The obtained OPSS-PEG-DOTA was used to conjugate gadolinium (Gd) to AuNS. Gadolinium chelated AuNS proved to have pent-modal imaging properties, including MRI, CT, OCT, RCM and two-photon luminescence (TPL) in tumor tissues. CT and MRI images also showed enhanced contrast in B16-F10 melanoma tumors in mice. Dai et al. first reported the preparation of AuNS on PAH and AuNP deposited PLA microcapsules with ultrasound imaging contrast.27 After intravenous injection, AuNS was able to enhance ultrasound imaging for New Zealand white rabbits' kidney in pulse-inversed harmonic imaging contrast mode. The effect of contrast enhancement could last for more than 5 min, while no arrhythmia or other side effects were observed. AuNS was further used as photothermal agents and 0.25 mg mL−1AuNS was able to depress more than 80% HeLa viability after irradiation under 808 nm laser at 4 W cm−2 for 10 min.
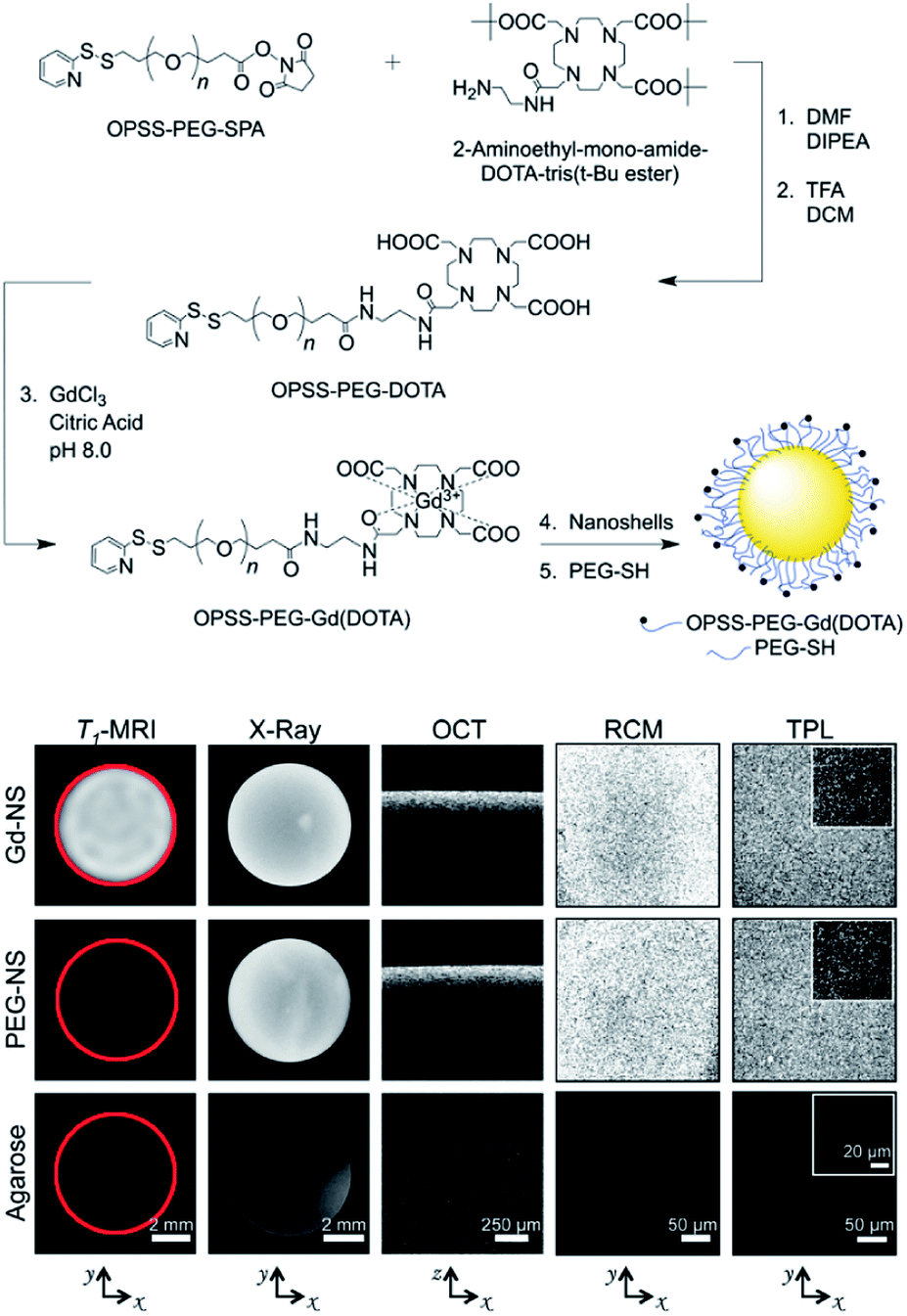 | ||
| Fig. 11 Scheme for OPSS-PEG-Gd (DOTA) synthesis and eventual conjugation to gold nanoshell surfaces. Gadolinium-nanoshells (Gd-NS) dispersed within agarose phantoms exhibited positive contrast across five imaging modalities: T1-weighted MRI, CT, OCT, RCM, and TPL. Phantoms with PEG-conjugated nanoshells (PEG-NS) offered no contrast under MR, similar to the 1% agarose control phantom, but comparable levels of contrast to Gd-NS with four other modalities. Red circles in T1-MRI column added post image acquisition to outline phantoms. Axes below indicate the plane across which phantom images were acquired within each column. Adapted from ref. 58. | ||
4. Conclusions
In summary, three methods, including AuNP surface coating, AuNP self-assembly, and gold deposition have been mainly developed for the preparation of polymer/AuNP hybrid nanoparticles, of which two basic strategies are often used for surface coating: (1) the ligand-exchange reaction and (2) the layer-by-layer electrostatic interactions. Thus, functional molecules including ATRP initiators can be introduced onto AuNP surface and the polymer/AuNP hybrid nanoparticles with more sophisticated structures and functions can be fabricated. Polymer/AuNP hybrids are ideal platforms for various cancer treatment modalities, the synergistic combination of chemo-photothermal therapy, and multiple imaging probes for cancer therapy. However, to precisely control the drug release (chemotherapy) and simultaneously achieve excellent photothermal properties (photothermal therapy) in one drug delivery vesicle is still challenging for realizing optimal synergistic effect in cancer therapy. In addition, the reproducible and simple large-scale synthesis and/or fabrication, in vivo biodegradability and biocompatibility, and the final fate of the polymer/AuNP hybrids need to be investigated thoroughly before they are potentially translated from bench to bedside.Acknowledgements
The authors are grateful for the financial support of the National Natural Science Foundation of China (21274086 and 21474061) and Shanghai Leading Academic Discipline Project (B202). Both Shanghai key lab of polymer and electrical insulation and the Instrumental Analysis Center of SJTU are also appreciated.References
- S. Lal, S. E. Clare and N. J. Halas, Acc. Chem. Res., 2008, 41, 1842–1851 CrossRef CAS PubMed.
- E. C. Dreaden, A. M. Alkilany, X. Huang, C. J. Murphy and M. A. El-Sayed, Chem. Soc. Rev., 2012, 41, 2740–2779 RSC.
- Y. C. Yeh, B. Creran and V. M. Rotello, Nanoscale, 2012, 4, 1871–1880 RSC.
- L. C. Kennedy, L. R. Bickford, N. A. Lewinski, A. J. Coughlin, Y. Hu, E. S. Day, J. L. West and R. A. Drezek, Small, 2011, 7, 169–183 CrossRef CAS PubMed.
- Y. Jin, Acc. Chem. Res., 2014, 47, 138–148 CrossRef CAS PubMed.
- Y. Zhong, C. Wang, L. Cheng, F. Meng, Z. Zhong and Z. Liu, Biomacromolecules, 2013, 14, 2411–2419 CrossRef CAS PubMed.
- R. Choi, J. Yang, J. Choi, E. K. Lim, E. Kim, J. S. Suh, Y. M. Huh and S. Haam, Langmuir, 2010, 26, 17520–17527 CrossRef CAS PubMed.
- J. Liu, C. Detrembleur, M. C. De Pauw-Gillet, S. Mornet, E. Duguet and C. Jerome, Polym. Chem., 2014, 5, 799–813 RSC.
- J. Liu, C. Detrembleur, B. Grignard, M. C. De Pauw-Gillet, S. Mornet, M. Treguer-Delapierre, Y. Petit, C. Jerome and E. Duguet, Chem.–Asian J., 2014, 9, 275–288 CrossRef CAS PubMed.
- J. Huang, K. S. Jackson and C. J. Murphy, Nano Lett., 2012, 12, 2982–2987 CrossRef CAS PubMed.
- F. Chen, S. Hou, Q. Li, H. Fan, R. Fan, Z. Xu, G. Zhala, X. Mai, X. Chen, X. Chen and Y. Liu, Anal. Chem., 2014, 86, 10021–10024 CrossRef CAS PubMed.
- M. S. Strozyk, M. Chanana, I. Pastoriza-Santos, J. Pérez-Juste and L. M. Liz-Marzán, Adv. Funct. Mater., 2012, 22, 1436–1444 CrossRef CAS.
- Q. Wei, J. Ji and J. Shen, Macromol. Rapid Commun., 2008, 29, 645–650 CrossRef CAS.
- Z. Zhang, J. Wang, X. Nie, T. Wen, Y. Ji, X. Wu, Y. Zhao and C. Chen, J. Am. Chem. Soc., 2014, 136, 7317–7326 CrossRef CAS PubMed.
- J. Li, J. Han, T. Xu, C. Guo, X. Bu, H. Zhang, L. Wang, H. Sun and B. Yang, Langmuir, 2013, 29, 7102–7110 CrossRef CAS PubMed.
- J. Hu, T. Wu, G. Zhang and S. Liu, J. Am. Chem. Soc., 2012, 134, 7624–7627 CrossRef CAS PubMed.
- J. Song, L. Cheng, A. Liu, J. Yin, M. Kuang and H. Duan, J. Am. Chem. Soc., 2011, 133, 10760–10763 CrossRef CAS PubMed.
- J. Song, J. Zhou and H. Duan, J. Am. Chem. Soc., 2012, 134, 13458–13469 CrossRef CAS PubMed.
- J. Song, L. Pu, J. Zhou, B. Duan and H. Duan, ACS Nano, 2013, 7, 9947–9960 CrossRef CAS PubMed.
- P. Taladriz-Blanco, N. J. Buurma, L. Rodriguez-Lorenzo, J. Perez-Juste, L. M. Liz-Marzan and P. Herves, J. Mater. Chem., 2011, 21, 16880–16887 RSC.
- Z. Sun, W. Ni, Z. Yang, X. Kou, L. Li and J. Wang, Small, 2008, 4, 1287–1292 CrossRef CAS PubMed.
- Y. Liu, X. Han, L. He and Y. Yin, Angew. Chem., Int. Ed., 2012, 51, 6373–6377 CrossRef CAS PubMed.
- D. Fava, M. A. Winnik and E. Kumacheva, Chem. Commun., 2009, 2571–2573 RSC.
- C. Wang, N. T. Flynn and R. Langer, Adv. Mater., 2004, 16, 1074–1079 CrossRef CAS.
- M. Lin, C. Guo, J. Li, D. Zhou, K. Liu, X. Zhang, T. Xu, H. Zhang, L. Wang and B. Yang, ACS Appl. Mater. Interfaces, 2014, 6, 5860–5868 CAS.
- J. Yang, J. Lee, J. Kang, S. Jae Oh, H. J. Ko, J. H. Son, K. Lee, J. S. Suh, Y. M. Huh and S. Haam, Adv. Mater, 2009, 21, 4339–4342 CrossRef CAS.
- H. Ke, J. Wang, Z. Dai, Y. Jin, E. Qu, Z. Xing, C. Guo, X. Yue and J. Liu, Angew. Chem., Int. Ed., 2011, 50, 3017–3021 CrossRef CAS PubMed.
- H. J. Yuan and C. M. Dong, Acta Polym. Sin., 2013, 3, 347–354 Search PubMed.
- H. Park, J. Yang, S. Seo, K. Kim, J. Suh, D. Kim, S. Haam and K. H. Yoo, Small, 2008, 4, 192–196 CrossRef CAS PubMed.
- S. M. Lee, H. Park and K. H. Yoo, Adv. Mater, 2010, 22, 4049–4053 CrossRef CAS PubMed.
- S. M. Lee, H. J. Kim, Y. J. Ha, Y. N. Park, S. K. Lee, Y. B. Park and K. H. Yoo, ACS Nano, 2013, 7, 50–57 CrossRef CAS PubMed.
- M. S. Yavuz, Y. Cheng, J. Chen, C. M. Cobley, Q. Zhang, M. Rycenga, J. Xie, C. Kim, K. H. Song, A. G. Schwartz, L. V. Wang and Y. Xia, Nat. Mater., 2009, 8, 935–939 CrossRef CAS PubMed.
- P. Shi, E. Ju, J. Ren and X. Qu, Adv. Funct. Mater., 2014, 24, 826–834 CrossRef CAS.
- Z. Wang, Z. Chen, Z. Liu, P. Shi, K. Dong, E. Ju, J. Ren and X. Qu, Biomaterials, 2014, 35, 9678–9688 CrossRef CAS PubMed.
- E. H. Jeong, J. H. Ryu, H. Jeong, B. Jang, H. Y. Lee, S. Hong, H. Lee and H. Lee, Chem. Commun., 2014, 50, 13388–13390 RSC.
- V. Garcia-Gradilla, S. Sattayasamitsathit, F. Soto, F. Kuralay, C. Yardımci, D. Wiitala, M. Galarnyk and J. Wang, Small, 2014, 10, 4154–4159 CAS.
- Y. Yang, Y. Hu, H. Du and H. Wang, Chem. Commun., 2014, 50, 7287–7290 RSC.
- Y. Cheng, T. L. Doane, C. H. Chuang, A. Ziady and C. Burda, Small, 2014, 10, 1799–1804 CrossRef CAS PubMed.
- J. Wang, G. Zhu, M. You, E. Song, M. I. Shukoor, K. Zhang, M. B. Altman, Y. Chen, Z. Zhu, C. Z. i. Huang and W. Tan, ACS Nano, 2012, 6, 5070–5077 CrossRef CAS PubMed.
- R. Vankayala, Y. K. Huang, P. Kalluru, C. S. Chiang and K. C. Hwang, Small, 2014, 10, 1612–1622 CrossRef CAS PubMed.
- L. Gao, R. Liu, F. Gao, Y. Wang, X. Jiang and X. Gao, ACS Nano, 2014, 8, 7260–7271 CrossRef CAS PubMed.
- Z. Shi, W. Ren, A. Gong, X. Zhao, Y. Zou, E. M. B. Brown, X. Chen and A. Wu, Biomaterials, 2014, 35, 7058–7067 CrossRef CAS PubMed.
- A. A. Zaki, D. Joh, Z. Cheng, A. L. B. De Barros, G. Kao, J. Dorsey and A. Tsourkas, ACS Nano, 2014, 1, 104–112 CrossRef PubMed.
- I. Miladi, C. Alric, S. Dufort, P. Mowat, A. Dutour, C. Mandon, G. Laurent, E. Bräuer-Krisch, N. Herath, J. L. Coll, M. Dutreix, F. Lux, R. Bazzi, C. Billotey, M. Janier, P. Perriat, G. L. Duc, S. Roux and O. Tillement, Small, 2014, 10, 1116–1124 CrossRef CAS PubMed.
- X. D. Zhang, J. Chen, Z. Luo, D. Wu, X. Shen, S. S. Song, Y. M. Sun, P. X. Liu, J. Zhao, S. Huo, S. Fan, F. Fan, X. J. Liang and J. Xie, Adv. Healthcare Mater., 2014, 3, 133–141 CrossRef CAS PubMed.
- H. C. Huang, S. Barua, D. B. Kay and K. Rege, ACS Nano, 2009, 10, 2941–2952 CrossRef PubMed.
- R. Huschka, J. Zuloaga, M. W. Knight, L. V. Brown, P. Nordlander and N. J. Halas, J. Am. Chem. Soc., 2011, 133, 12247–12255 CrossRef CAS PubMed.
- R. Huschka, A. Barhoumi, Q. Liu, J. A. Roth, L. Ji and N. J. Halas, ACS Nano, 2012, 6, 7681–7691 CrossRef CAS PubMed.
- E. R. Figueroa, A. Y. Lin, J. Yan, L. Luo, A. E. Foster and R. A. Drezek, Biomaterials, 2014, 35, 1725–1734 CrossRef CAS PubMed.
- X. Y. Wan, L. L. Zheng, P. F. Gao, X. X. Yang, C. M. Li, Y. F. Li and C. Z. Huang, Sci. Rep., 2014, 4, 4529 Search PubMed.
- J. Nam, W. G. La, S. Hwang, Y. S. Ha, N. Park, N. Won, S. Jung, S. H. Bhang, Y. J. Ma, Y. M. Cho, M. Jin, J. Han, J. Y. Shin, E. K. Wang, S. G. Kim, S. H. Cho, J. Yoo, B. S. Kim and S. Kim, ACS Nano, 2013, 7, 3388–3402 CrossRef CAS PubMed.
- C. Xu, G. A. Tung and S. Sun, Chem. Mater., 2008, 20, 4167–4169 CrossRef CAS PubMed.
- P. Yan, N. Zhao, H. Hu, X. Lin, F. Liu and F. J. Xu, Acta Biomater., 2014, 10, 3786–3794 CrossRef CAS PubMed.
- D. Huo, J. Ding, Y. X. Cui, L. Y. Xia, H. Li, J. He, Z. Y. Zhou, H. W. Wang and Y. Hu, Biomaterials, 2014, 35, 7032–7041 CrossRef CAS PubMed.
- T. Liu, I. Kempson, M. de Jonge, D. L. Howard and B. Thierry, Nanoscale, 2014, 6, 9774–9782 RSC.
- P. Huang, J. Lin, W. Li, P. Rong, Z. Wang, S. Wang, X. Wang, X. Sun, M. Aronova, G. Niu, R. D. Leapman, Z. Nie and X. Chen, Angew. Chem., Int. Ed., 2013, 52, 1–7 CrossRef.
- X. Sun, X. Huang, X. Yan, Y. Wang, J. Guo, O. Jacobson, D. Liu, L. P. Szajek, W. Zhu, G. Niu, D. O. Kiesewetter, S. Sun and X. Chen, ACS Nano, 2014, 8, 8438–8446 CrossRef CAS PubMed.
- A. J. Coughlin, J. S. Ananta, N. Deng, I. V. Larina, P. Decuzzi and J. L. West, Small, 2014, 10, 556–565 CrossRef CAS PubMed.
| This journal is © The Royal Society of Chemistry 2015 |