
 Open Access Article
Open Access Article This Open Access Article is licensed under a
This Open Access Article is licensed under a Creative Commons Attribution 3.0 Unported Licence
Crosslinked reduced graphene oxide/polymer composites via in situ synthesis by semicontinuous emulsion polymerization†
D.
Spasevska
a,
G. P.
Leal
b,
M.
Fernández
b,
J. Blazevska
Gilev
a,
M.
Paulis
b and
R.
Tomovska
*bc
aFaculty of Technology and Metallurgy, University of Cyril and Methodius, Rudjer Boskovic 16, 1000 Skopje, R. Macedonia
bPOLYMAT, Facultad de Ciencias Químicas, University of the Basque Country UPV/EHU, Joxe Mari Korta Zentroa, Tolosa Etorbidea 72, Donostia-San Sebastián 20018, Spain. E-mail: radmila.tomovska@ehu.es
cIKERBASQUE, Basque Foundation for Science, Maria Diaz de Haro 3, Bilbao 48013, Spain
First published on 28th January 2015
Abstract
This work presents the in situ synthesis of crosslinked polymer/graphene nanocomposites by seeded semicontinuous emulsion polymerization. The OH functionalities on the rGO platelets and in the polymer chains (introduced by the functional monomer hydroxylethyl methacrylate) were used as reactive sites, linked by the presence of NCO terminated polyurethane prepolymer (PU). The reaction was performed in semicontinous mode, using rGO and PU dispersed in water as a seed, whereas the monomers preemulsion was fed slowly. The hybrid latexes were kinetically stable enough to produce composite films by water evaporation, in which rGO platelets were uniformly dispersed and incorporated in a permanent way in the polymer matrix. The composites have a highly crosslinked structure, the degree of which depends on the loading of rGO and PU e.g., NCO/OH ratio. The electrical conductivity of the composites depends highly on the degree of crosslinking and the morphology, thus aligned platelets parallel to the top film surface are the most prospective morphology for the electrical conductivity. Determination of viscoelastic properties has shown that the composites are stiffer and contain smaller amounts of the mobile neat polymer phase with increasing content of rGO, which agrees well with an increase in their crosslinking.
Introduction
Versatility of polymers for application in almost all spheres of human life places new demands for improvement of their properties and performance. They are required to present very good mechanical properties, stability at extreme conditions, possibilities of self-healing, sensitivity at different atmospheric conditions, electrical or thermal conductivity, etc. One of the possible ways to expand the properties of polymers is by the introduction of suitable fillers that will extend the application of the resulting composite materials. Different shape and morphology nanofillers have been incorporated into polymer matrices, such as spherical nanoparticles: silica,1–3 titania,4,5 noble metals,6etc.; platelet-like two dimensional fillers: clays,7,8 or graphene;9–34 and one dimensional fillers: nanotubes or fibers.35One of the recently most investigated nanofillers with a multifunctional nature that offers the introduction of a wide variety of properties into polymers is graphene. It has been used to introduce electrical conductivity,9–21,26,31–33 thermal stability,21–23,26,30 flame retardancy,28,29 or sensing properties,24 or to improve the mechanical properties,10,14,17–21,26,30,32,34etc. In the recent literature various techniques are reported to be suitable for the preparation of polymer/graphene composites, such as simple physical mixing of graphene and the polymer in solution,12,14,19 emulsion9,10,13,15–20 or bulk (by melting),26–28 or in situ techniques, when the polymerization is performed in the presence of graphene in solution, in dispersed media or in bulk.9,11,23,29–34
The in situ polymerization techniques are rather challenging regarding the reproducibility and the control of the product microstructure and properties. They offer the possibility to synthesize stable nanocomposites with improved mechanical and/or thermal properties as a result of establishing a more intimate contact between the filler and the polymer.31 However, the interactions reported to be created during in situ synthesis, are not controlled in most of the cases and occur spontaneously,9,31–33 which could influence negatively the composite properties. For example, covalent bonds established between the polymer and graphene have been demonstrated in some works dealing with in situ emulsion polymerization of various monomers in the presence of graphene,9,31 although no details on the bonding were provided. Significant decrease in the composite's electrical conductivity was accounted on the change of sp2 hybridization of carbon into sp3 carbons in the graphene structure.9,31
In order to design composite materials made of covalently bonded graphene platelets and polymer chains, in which the conductivity of the platelets will not be altered significantly during synthesis, we performed in situ polymerization in which we created reactions sites by introduction of suitable functionalities onto both components (rGO and polymer). This allowed bonding between the polymer chains and graphene platelets to take place in a controlled way and to decrease the possibilities of spontaneous bonding that alter the rGO structure. This was carried out by introduction of OH functionalities in polymer and aliphatic polyurethane (PU) prepolymer with free isocyanate groups on both chain ends, which establish covalent bond with both OH of polymer and of reduced graphene oxide. The whole procedure was performed in emulsion, using water as a reaction medium, which is an additional advantage of this technique and contributes toward the development of environmental friendly synthesis methods.
Recently, we have performed a similar study using the advantage of the proposed NCO–OH chemistry by simple blending of emulsion acrylate polymers and rGO in presence of a polyisocyanate crosslinker.10 The synthesis of highly stable hybrid polymer films, electrically conductive and with improved mechanical properties was reported. However, the hybrid water dispersion containing polymer nanoparticles and rGO platelets destabilized fast after addition of crosslinker, since a kind of coagulation occurred during forming of composite nanostructures. This limits their application as electrically conductive and protective coatings. That is why in this work we propose the creation of composite nanoparticles in situ by polymerization in emulsion in order to obtain stable hybrid aqueous dispersions, suitable for composite film preparation on various substrates.
Up to the best of authors' knowledge this is first time to report the use of seeded semicontinuous emulsion polymerization in situ for the synthesis of polymer/graphene composites and the first report on introducing controlled reaction between both components. As the modified rGO platelets containing NCO reactive groups on their surface are used as a seed, we expect that the polymer nanoparticles will start to grow from this reaction site. The advantage of the semicontinuous process is that the reaction is performed under low monomer concentration, which allows better control of the composite nanoparticles structure and creation of composite polymer/graphene nanoparticles instead of separate polymer particles in rGO presence. The covalent bonding between them brings stability, electrical conductivity and improves the mechanical performance of the nanocomposite. As well, semicontinous emulsion polymerization is an industrially significant process, which makes synthesis of graphene emulsion composites one step closer to possible commercialization.
Experimental part
Materials
Technical-grade monomers methyl methacrylate (MMA, Quimidroga), butyl acrylate (BA, Quimidroga) and 2-hydroxy ethyl methacrylate (HEMA, Fluka) were used without further purification. Sodium dodecyl sulfate (SDS, Aldrich) and polyvinylpyrrolidone (PVP) were used as received, as surfactants. Potassium peroxodisulfate (KPS) (Fluka analytical) was used as an initiator. The aliphatic isocyanate terminated polyurethane (PU) prepolymer Incorez 701 (PU) (Incorez Ltd.), with an equivalent weight of 1050 g per equivalent, was used without further purification. Graphene oxide (GO) aqueous dispersion (Graphene Supermarket) containing 4 g L−1 GO, 60% monolayer and 400 nm average platelets diameter was used for preparation of reduced graphene oxide (rGO). Hydrazine monohydrate (Aldrich) was used as received for reduction of GO. Tetrahydrofuran (THF; Scharlau) and methanol (Scharlau) were used as solvents. Oxygen-free-grade nitrogen was used for purging the feed. Double deionized water was used throughout experimentation.GO reduction
The procedure of GO reduction by hydrazine in the presence of PVP is explained elsewhere.9 Shortly, the reaction was performed at 90 °C over 3 h and the hydrazine/GO reactants ratio was 5![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) :1 by weight. After the reduction the dispersion was dialyzed using dialysis membrane Spectral/Por (Spectrumlabs) with MWCO: 12–14000 Daltons. This way the side products of the reaction and the extra PVP were removed and neutral pH was achieved. The concentration of rGO in the dispersion after the reduction was always slightly higher than 2 wt%, as determined by elemental analysis. The platelets were mostly one to two layers thick, as determined by AFM analysis.9
:1 by weight. After the reduction the dispersion was dialyzed using dialysis membrane Spectral/Por (Spectrumlabs) with MWCO: 12–14000 Daltons. This way the side products of the reaction and the extra PVP were removed and neutral pH was achieved. The concentration of rGO in the dispersion after the reduction was always slightly higher than 2 wt%, as determined by elemental analysis. The platelets were mostly one to two layers thick, as determined by AFM analysis.9
In situ polymerization
In situ seeded semicontinuous emulsion polymerizations of MMA/BA/HEMA (49.5/49.5/1 wt% ratio) in the presence of rGO (0.62–1 wt% with respect to monomers) and PU (0–20 wt% with respect to the monomers) were carried out. The PU was dispersed in the PVP stabilized aqueous dispersion of rGO, where 1 wt% of SDS (with respect to monomers) was added to assure the colloidal stability of the dispersion. This colloidal dispersion was placed in a reactor after being mixed for 30 minutes and used as seed in the semicontinuous emulsion polymerization of MMA/BA/HEMA. The monomers were fed as a pre-emulsion containing 1 wt% SDS (with respect to monomers) and water (see Table 1). The feeding of the monomers started when 70 °C were reached in the reactor and the initiator KPS was added as a shot. After 180 minutes all components were introduced in the reactor and left there to react additional 60 minutes. The formulation used for the synthesis of hybrid latex containing 10 wt% PU and 0.75 wt% rGO is presented in Table 1.Film formation
The composite films were prepared from the hybrid latexes by water evaporation under controlled conditions (25 °C and 80% humidity) in Temperature and Humidity Chamber (Espec, SH-641). The hybrid latexes were casted into 5.5 × 2.5 mm silicone moulds and allowed to form a film for 24 hours.Characterization
The monomer conversion was determined gravimetrically. The mean particle diameters of the hybrid latexes were measured by using dynamic light scattering (Zetasizer Nano Z, Malvern Instruments). The samples were prepared by diluting one drop of latex in deionized water. The particle diameters reported are the z-average of two measurements. The average macroscopic gel content was determined gravimetrically by Soxhlet extraction under THF reflux. 10–15 latex drops were placed on glass fiber square pads of weight W1 and they were dried at 60 °C for 24 h (weight W2). Then, the samples were subjected to 24 h Soxhlet extraction under THF reflux and after dried overnight (weight W3). The gel content was calculated as the ratio of the dry polymer remaining after the extraction and the initial amount of dry polymer, according to the expression (1): | (1) |
The degree of equilibrium swelling was determined in the following way. Weighted amounts of the composite films (weight Wi) were placed in THF solvent at 25 °C. After 24 h they were removed from the solvent, dried quickly with an absorbent paper, weighted (weight Wj) and returned to the solvent. The procedure was repeated until constant Wj was obtained. The degree of the equilibrium swelling was obtained as a ratio of Wj/Wi.
The electrical conductivity was measured by a four point probe (Miller Inc.m model FP500). The measurements were performed in several points of the film on top and bottom surfaces of the composite film. The presented conductivities are the average of 5 measurements.
FT-IR spectra were obtained using ALPHA spectrometer coupled to a Golden Gate single reflection attenuation total reflection (ATR) unit.
The morphology of the composite films was investigated by a scanning electron microscope (SEM) (Quanta FEG 250, high vacuum mode at 5 KV). Samples were scanned without previous sputtering of a metal on their surface. Cross-sectional imaging was taken after breaking the samples under liquid nitrogen.
Wide-angle X-ray diffraction (WAXD) analyses of the hybrid films were performed on a D8 Advance (Bruker) (CuKα radiation with λ = 0.154056 nm). The range of the diffraction angles was 2θ = 2–50° at a scanning rate of 0.05° × (5 s)−1.
Dynamic mechanical thermal analysis (DMTA) measurements were conducted on a Tritec 2000 DMA manufactured by Triton Technology (UK). Thermal scans were performed at a frequency of 1 Hz with a heating rate of 4 °C min−1, in order to measure the storage (E′), loss modulus (E′′) and the loss tangent (tanδ) as a function of temperature. All samples were run in single cantilever bending mode with a displacement of 0.005 mm and a length between the clamps of 2 mm.
Results and discussions
The in situ synthesis of the polymer/graphene composites resulted in full monomer conversion and formation of kinetically stable hybrid latexes (sedimentation started to take place only after few weeks). After almost one year of storage, the hybrid latex obtained without PU shows phase separation, whereas the hybrid latexes containing PU beside a small sediment, keep the colloidal dispersion appearance (the sedimentation was in the range of 3–6 wt%). In the ESI† the mean particle diameters of just synthesized latexes are compared with those of the latexes stored for almost one year (Table S1†). The results clearly indicate that most of the composite particles remained stabilized in the aqueous dispersions and that the small sedimented part was redispersible by hand shaking. Furthermore, the similarity between the particle diameters of just synthesized composite latex and those obtained after storage confirm the stability of the in situ synthesized hybrids, even if a slight aggregation cannot be discarded.FTIR spectra of the composites (presented in ESI, Fig. 1s†) show that NCO groups from the PU were completely consumed as the NCO characteristic vibration centred at around 2265 cm−1 was not present in the spectra of composite materials.
The gel content (THF insoluble fraction of the composites) for different rGO amounts and various PU loadings, determined by Soxhlet extraction under THF reflux is presented in Table 2. It is worth noting that after the extraction there were no rGO platelets in the solution even after months of storage, meaning that all rGO platelets were strongly bonded into the composite structure, which indicates that the crosslinking reactions between rGO and polymer took place.
| PU content (wt%) | rGO content (wt%) | Gel content (wt%) | Swelling at 25 °C | Electrical conductivity (S m−1) |
|---|---|---|---|---|
| 0 | 0.62 | 14 | 8.7 | (2.68 ± 0.220) × 10−4 |
| 5 | 0.62 | 63 | 4.9 | (2.20 ± 0.004) × 10−3 |
| 10 | 0.62 | 48 | 6.8 | (0.66 ± 0.110) × 10−3 |
| 20 | 0.62 | 47 | 7.0 | (1.04 ± 0.001) × 10−3 |
| 10 | 0.75 | 53 | 6.5 | (3.00 ± 0.006) × 10−2 |
| 10 | 1.00 | 65 | 5.0 | (2.96 ± 0.011) × 10−2 |
Table 2 and Fig. 1 show that for constant rGO loading addition of 5 wt% PU with respect to monomers increased significantly gel content from 14 to 63 wt%, achieved likely throughout crosslinking of rGO, PU and MMA/BA/HEMA polymer chains. Further increase in PU content to 10–20 wt% with respect to monomer dropped the gel content (Fig. 1). It is well known that the reactivity of the second NCO group in the polyurethane prepolymer is decreased after the first NCO has been converted to urethane structure (–NH–CO–O–).36 In a high surplus of NCO groups, OHs from rGO and polymer will be preferably spent in the reaction with the first NCOs from PU, decreasing the overall crosslinking efficiency and therefore the gel content. As NCO conversion was full, it is quite possible that the rest of isocyanate functionalities are consumed by reactions with water and if eventually not included in the composite structures, they could decrease the composite performance, due to the phase separations.
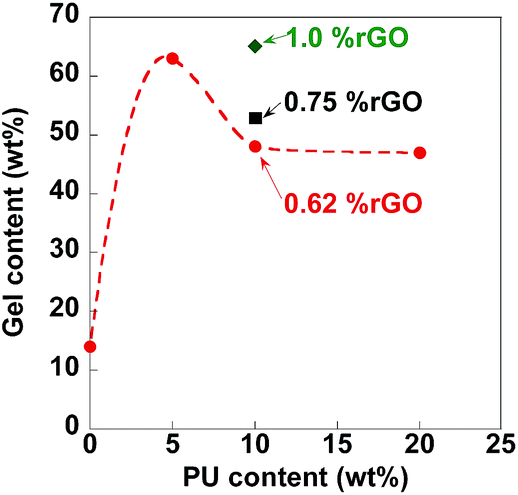 | ||
| Fig. 1 Gel fraction of nanocomposites as a function on PU content; for constant PU content of 10%, the rGO contents for the different point are placed next to each point. The dished line is guide to the eye. | ||
Crosslinking degree increases with the rGO contents (0.62–1 wt%) (Table 2, Fig. 1b), due to more OH reaction sites available per constant NCOs.
The composite films were prepared from the hybrid latexes by water evaporation under controlled conditions (20 °C and 80% relative humidity). When the composite films were placed in THF solvent at room temperature (25 °C), they swelled in the solvent, which resulted in a volume increase and confirms that the composites have a crosslinked structure. The degree of equilibrium swelling at 25 °C is shown in Table 2.
The less crosslinked film (0% PU) presents the highest swelling degree (8.7). In comparison to the blank poly(MMA/BA/HEMA) film with a degree of swelling of 24.0 (and <5% gel content) this composite films obtained by in situ reaction in presence of rGO platelets but without any addition of PU already show certain crosslinking as significant decrease of the swelling was measured. This means that the covalent bonding between the rGO platelets and polymer chains occurred, which is in accordance with our previous results9 and literature reports31 obtained under similar conditions. Addition of PU introduces an increase of the crosslinking degree in the composites, as the measured swelling values decrease, which agrees well with the gel content values (Table 2).
Based on these findings we have proposed a mechanism of formation of composite particles, shown in Scheme 1.
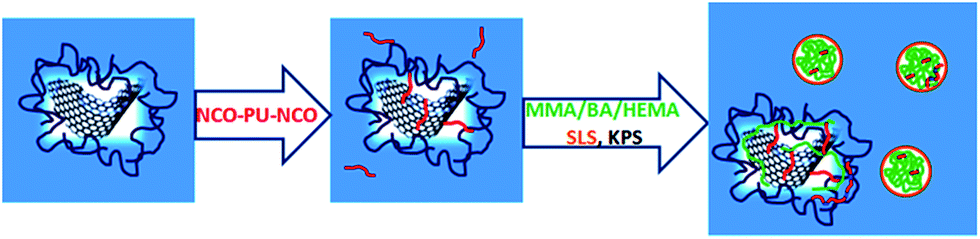 | ||
| Scheme 1 rGO/PU/polymer reactions. | ||
When PU comes in contact with rGO platelets, a kind of in situ surface modification of the platelets occurs throughout NCO reaction with OH, as presented in Scheme 1. In this way the platelets are functionalized with PUs that still contain pendant reactive NCO functional groups. The monomers were fed in pre-emulsion, which means that surfactant (SDS) and water soluble initiator (KPS) were introduced in the dispersion. This can give rise to homogeneously nucleated polymer particles (growing of polymer chains in aqueous phase by free radical polymerization, their precipitation and stabilization by surfactant). As polymer particles are functionalized with OH functionalities, in the course of reaction they react with PU or rGO/PU by addition polymerization. In this way the crosslinking occurred, resulting in the formation of composite particles. The other possibility is the reaction between NCO functionalities at rGO platelets with the OH group of HEMA monomer and subsequent their incorporation in the growing acrylic chains. This means that polymer particle grows from the surface of rGO platelets, which is another route of composite particle nucleation. Additionally, under higher PU contents, the reaction of polycondensation becomes significant (PU pre-polymer reacts with water) producing extending of the PU chains that either can be incorporated in the composite structure or coexist in the dispersion as a separate polyurethane phase reach with polyurea groups. By controlling of NCO/OH ratio the presence of all of these structures can be controlled, as demonstrated by different gel contents and degrees of crosslinking.
The electrical conductivities of the composite films are presented in Table 2. In situ reaction performed in presence of 0.62 wt% of rGO without PU added resulted in composite films with the lowest conductivity measured in this work (2.68 × 10−4 S m−1). In comparison to a neat polymer insulating film (<10−6 S m−1), this is already a significant enhancement; however the addition of PU during in situ reaction in presence of rGO further increases the electrical conductivity up to two orders of magnitude. The composite film containing 10 wt% PU and 0.75 wt% rGO presents the best electrically conductive properties (3 × 10−2 S m−1). The difference in the conductivities comes likely due to preferably established covalent bonds between the OH already present in the rGO structure rather than the spontaneous incorporation in the rGO structures by creation of a new sp3 hybridized C atoms. In the reactions performed without PU, covalent bonding occurs in this way, decreasing the overall conductivity of the composites. The best conductive properties of the composite film containing 10 wt% PU and 0.75% rGO are an indication that these amounts of PU and rGO provide a kind of optimum NCO/OH ratio. Further increase in rGO content to 1 wt% for the same PU concentration did not introduce the expected augmentation of conductivity. As the gel content was increased for 1 wt% rGO, this means that not only the amount of rGO will influence the conductivity, but the microstructure and the morphology.
In Fig. 2 the morphology of the composite films is presented. The SEM images of the cross-sectional area of the films broken under liquid nitrogen are shown. The images were obtained without covering the surface with conductive metal film, demonstrating that all composites are conductive. The white/gray rGO platelets are well distributed in the black polymer matrix.
 | ||
| Fig. 2 SEM cross-sectional images of composite films containing (a) 5 wt% PU and 0.62 wt% rGO; (b) 10 wt% PU and 0.75 wt% rGO; and (c) 10 wt% PU and 1 wt% rGO. | ||
Fig. 2a shows the morphology of composite film that contains 5 wt% PU and 0.62 wt% rGO. The rGO platelets are quite randomly dispersed in the polymer matrix, but there are neat polymeric areas of around 1 μm. According to Table 2, this composite is highly crosslinked, which is likely the reason for the random orientation, because the crosslinked structures decrease the mobility of the rGO platelets and prevents their organization in one direction during water evaporation and composite film formation. Fig. 2b shows the composite that contains 10 wt% PU and 0.75 wt% rGO, for which the maximum conductivity was achieved. Crosslinked fraction in this material is lower (53%). The rGO platelets are very well organized in direction perpendicular to the view (since we see the cross-sectional area, the platelets are organized in direction parallel to the top surface of the film) and no aggregation is visible. The well defined polymeric areas of around 300 nm are surrounded by the platelets. This morphology seems to be prosperous for the electrically conductive properties, as already has been reported.9 Further increase of rGO loading to 1 wt%, as shown in Fig. 2c, increases the gel content that gives rise to composites in which the platelets are oriented randomly, whereas the neat polymeric areas are even smaller. It seems that if the higher electrical conductivity is the goal, the compromise should be established between the crosslinking structure (PU content) and the filler loading.
In order to check the exfoliation of the rGO platelets in the polymer matrix XRD analysis were performed. The XRD spectra for the composites prepared in situ with the same amount of rGO (0.62 wt%) and different amounts of PU (0–20%) are presented in Fig. 3 and compared with the blend from the same polymer with graphite. As it can be seen, the peak corresponding to the stacked structure of the graphite (26.6°) disappears completely in the composites prepared in situ, showing the exfoliated structure of the rGO in the composite films. This exfoliation is also maintained even at 1 wt% of rGO (see Fig. 4). The peaks appearing at 2.7°, 5.2° and 7.8° are SDS crystallization peaks,37 which has perhaps migrated from the particles surface to the film surface during film formation process. In the case of the composites with the same PU content (10 wt%) and different rGO contents (0.62–1.0 wt%) (Fig. 4), these SDS crystallization peaks seem to disappear as the rGO content is increased. This decrease in the SDS migration during film formation can be produced by the more tortuous path generated in the presence of higher amounts of rGO.
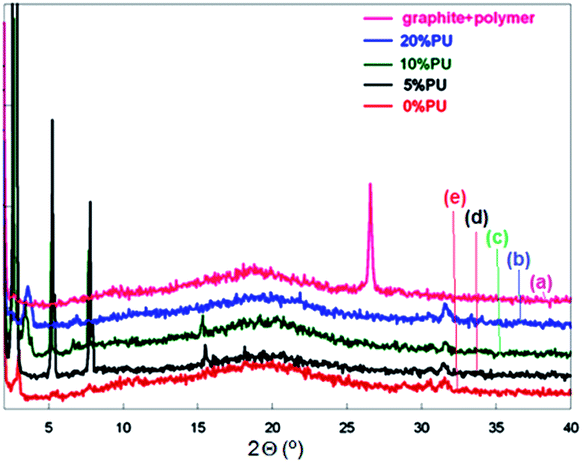 | ||
| Fig. 3 Comparison of WAXD patterns of composite film containing 1% graphite (a) and composite films containing 0.62 wt% rGO and different amounts of PU: (b) 20 wt%; (c) 10 wt%; (d) 5 wt%; and (e) 0 wt%. | ||
 | ||
| Fig. 4 Comparison of WAXD patterns of composite films containing 10% PU and different amounts of rGO: (a) 0.62 wt% (b) 0.75 wt%; and (c) 1 wt%. | ||
Finally we would like to comment on the peak at 3.4–3.6° for the composites with 0.62 wt% of rGO and 10 and 20 wt% of PU. It is a peak that appears for the combination of the highest PU and lowest rGO contents. No clear explanation can be given up to the moment for this peak, but the high excess of PU in relation to rGO induces lack of linkage between them. Under these conditions the reaction between PU and water by polycondensation could become significant, creating extension of the PU chains by polyurea groups. These PU chains could appear as additional softer phase in the composites or be attached to the rGO platelets or poly(MMA/BA/HEMA) chains.
Viscoelastic properties of the composites, presented in Fig. 5 and 6, were obtained by DMTA measurements. In Fig. 5a and b, the influence of PU content on storage modulus (Fig. 5a) and on tanδ (Fig. 5b) is shown, comparing the nanocomposites with neat polymer and with polymer containing the same amount of rGO, but no PU (the intersections of the Fig. 5a and b). The neat polymer presents lower modulus, especially highlighted at higher temperatures and liquid like behavior induced by temperature, which is clear from the significant drop of the modulus and abrupt increase in tanδ. This behavior is not present in the nanocomposites. The addition of rGO allowed to retain a higher modulus even at temperature above the glass transition. Actually, it is observed a combination of the reinforcement effects from the filler and from the high croslinking structure Fig. 5a shows that obviously in the glassy state the PU content influences significantly the properties, and the reinforcing effect is decreased by increasing the PU content. However, in the rubbery plateau region the properties are the same for all films as they all have the same amount of rGO (0.62%), and the rubbery modulus increment is mainly due to the fact that rGO has a much higher modulus than the matrix, showing that PU did not influence them. It is worth noting that the sample containing 20% PU show decreasing modulus and lowering the reinforcing effect. As previously discussed, in the presence of high amount of PU and NCO functionalities they react with water by polycondensation and form additional phase containing a plenty of polyurea functionalities. This is demonstrated in Fig. 5b, where the presence of additional small peak for 20% PU (Tg ∼ −52 °C) reveals the presence of a second softer phase in this nanocomposite.
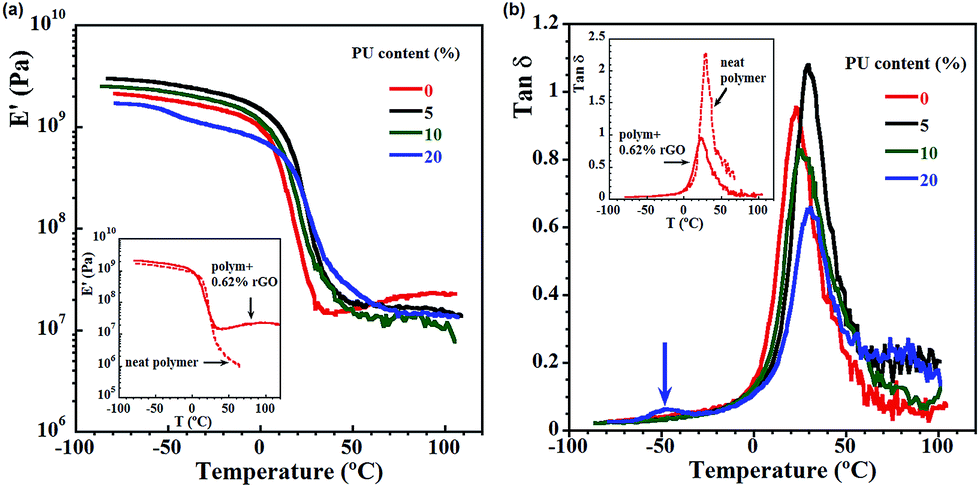 | ||
| Fig. 5 Viscoleastic properties of the composites containing 0.62 wt% rGO with respect to monomers and different amounts of PU: (a) dependence of storage modulus on temperature; and (b) dependence of tanδ on temperature. | ||
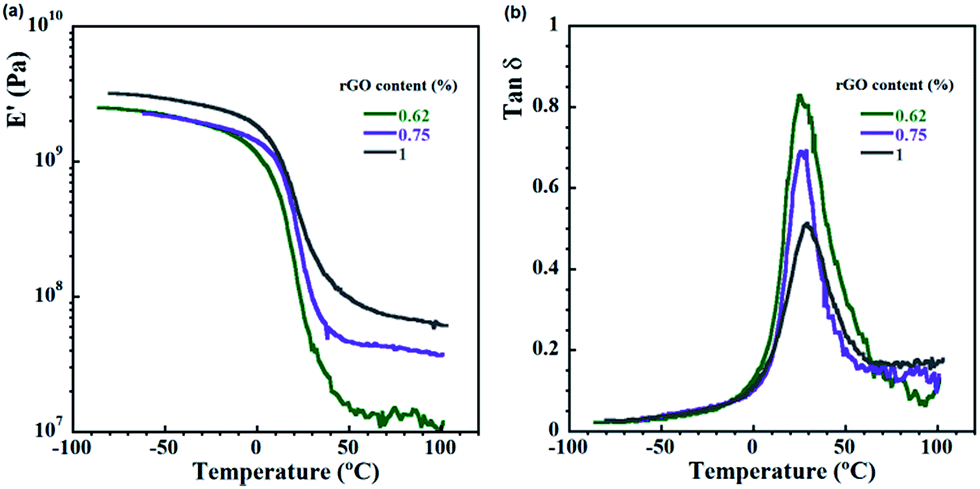 | ||
| Fig. 6 Viscoleastic properties of the composites containing 10 wt% PU with respect to monomers and different amounts of rGO: (a) dependence of storage modulus on temperature; and (b) dependence of tanδ on temperature. | ||
tanδ is related to the relaxation behaviour of local chain segments occurring in the polymer. For nanocomposites, the molecular movement at the interphase contributes to the value of the tanδ that enables to estimate the bonding between the interphase of the matrix and the rGO platelets. The presence of PU apparently improves the interactions between the matrix and the filler resulting in the expected increase of Tg. However, the Tg's shift depends on the PU content and the strongest effect is observed for composites containing 5% PU as Tg increased from 22.3 °C for 0% PU to 30 °C for the nanocomposite. Further increase of PU content didn't bring expected increase in Tg values. Since these systems are rather complicated, it can be assumed that the Tg transition is reduced due to a combination of factors. In fact, the reduction of crosslinking density as the PU content increases could play a role, and as it was mentioned, in presence of higher amounts of PU the polycondensation reaction of prepolymer and water occurred, giving a rise to the additional soft phase within the composite (for 20 wt% PU) or presence of pendant PU/polyurea groups (10 wt% PU) that acts as a plasticizers and also can decreased the expected Tg.
In Fig. 6a and b the influence of rGO content on the storage modulus and tanδ is presented. The storage modulus increases with the rGO content and the reinforcement effect becomes even more pronounced in the rubbery state of the nanocomposites, where increasing rGO content from 0.62 to 1 wt% increases the modulus one order of magnitude. The height of tanδ peak decrease significantly (Fig. 6b) with increasing rGO content, revealing that during the glass transition the amount of mobile polymer phase is fewer. This is in agreement with the crosslinking degree (Table 2) and the morphology (Fig. 2), as by increasing it the mobility in the glass transition region of the polymer phase is reduced. This results in a slight augmentation of glass transition temperature, as the Tg values moved from 24°, to 26° and 28 °C respectively for 0.62, 0.75 and 1% rGO. In comparison with the Tg of reactive composite blends in similar system (20.6 °C for 1 wt% rGO),10 obviously in situ synthesis presented in this work improved bonding between the phases.
Conclusions
Creation of reaction sites by introduction of appropriate functionalities during in situ seeded emulsion polymerization of MMA/BA/HEMA in presence of rGO platelets and PU prepolymer containing isocyanate functionalities resulted in synthesis of highly crosslinked, electrically conductive polymer composites. The stability of the composites has been demonstrated by solubility test in THF, which proved that the rGO platelets are included in the polymer structure in a permanent way. The crosslinking degree has shown to depend on the amount of the rGO and PU added, i.e. NCO/OH ratio. As well this ratio has shown to influence a lot the morphology of the film, the electrical conductivity and finally the mechanical properties of the composite films. Increasing the crosslinking in the composites up to some content gives rise to nice rGO distribution in polymer matrix, and incorporation of the platelets in an organized manner (parallel to the top surface of the composite films). This organized morphology resulted in the highest achieved electrical conductivity and improved mechanical properties. Further increase of PU content induced side reaction of polycondensation (PU with water), dropped the crosslinking degree, the conductivity and the reinforcing effect. The rGO loading in the investigated range (0.62–1 wt%) positively influenced the properties, thus the crosslinking degree and the reinforcement effect increased monotonically, whereas the conductivity increased up to the 0.75 wt% filler loading, after which it slightly decreased. This effect was limited by the amount of PU, as it seems that always a compromise should be found between the amounts of OH and NCO in the system for optimal effects.Acknowledgements
The financial support of the Basque Government (GV IT373-10), NATO (SfP 984399), Diputación Foral de Gipuzkoa (Exp. 55/14) and University of the Basque Country UPV/EHU (UFI11/56) is gratefully acknowledged.References
- E. Reynaud, C. Gauthier, G. Vigier and J. Varlet, Polymer, 2001, 42, 8759 CrossRef CAS.
- D. A. Savin, J. Pyun, G. D. Patterson, T. Kowalewski and K. Matyjaszewski, J. Polym. Sci., Part B: Polym. Phys., 2002, 40, 2667 CrossRef CAS.
- K. González-Matheus, G. P. Leal and J. M. Asua, Part. Part. Syst. Charact., 2014, 31, 94 CrossRef.
- L. M. Hamming, R. Qiao, P. B. Messersmith and L. C. Brinson, Compos. Sci. Technol., 2009, 69, 1880 CrossRef CAS PubMed.
- Q. F. Xu, Y. Liu and F. J. Lin, ACS Appl. Mater. Interfaces, 2013, 5, 8915–8924 CAS.
- G. Walters and I. P. Parkin, J. Mater. Chem., 2009, 19, 574 RSC.
- A. Bonnefond, M. Paulis, S. A. F. Bon and J. R. Leiza, Langmuir, 2013, 29, 2397 CrossRef CAS PubMed.
- A. Bonnefond, M. Micusik, S. A. F. Bon, M. Paulis and J. R. Leiza, Colloid Polym. Sci., 2013, 291, 167 CAS.
- A. Arzac, G. P. Leal, R. Fajgar and R. Tomovska, Part. Part. Syst. Charact., 2014, 31, 143 CrossRef CAS.
- D. Spasevska, V. Daniloska, G. P. Leal, J. Blazevska Gilev and R. Tomovska, RSC Adv., 2014, 4, 24477 RSC.
- L. Yang, J. Kong, W. A. Yee, W. Liu, S. L. Phua, C. L. Toh, S. Huang and X. Lu, Nanoscale, 2014, 2, 4968 Search PubMed.
- V. H. Pham, T. V. Cuong, T. T. Dang, S. H. Hur, B. S. Kong, E. J. Kim, E. W. Shin and J. S. Chung, J. Mater. Chem., 2011, 21, 11312 RSC.
- N. Yousefi, M. M. Gudarzi, Q. Zheng, S. H. Aboutalebi, F. Sharif and J. K. Kim, J. Mater. Chem., 2012, 22, 12709 RSC.
- X. Y. Qi, D. Yan, Z. Jiang, Y. K. Cao, Z. Z. Yu, F. Yavari and N. Koratkar, ACS Appl. Mater. Interfaces, 2011, 3, 3130 CAS.
- E. Tkalya, M. Ghislandi, A. Alekseev, C. Koning and J. Loos, J. Mater. Chem., 2010, 20, 3035 RSC.
- Y. V. Syurik, M. G. Ghislandi, E. E. Tkalya, G. Paterson, D. Mc Grouther, O. A. Ageev and J. Loos, Macromol. Chem. Phys., 2012, 213, 1251 CrossRef CAS.
- A. Noel, J. Faucheu, J.-M. Chenal, J.-P. Viricell and E. Bourgeat-Lami, Polymer, 2014, 55, 5140 CrossRef CAS.
- A. Noel, J. Faucheu, M. Rieu, J.-P. Viricelle and E. Bourgeat-Lami, Compos. Sci. Technol., 2014, 95, 82 CrossRef CAS.
- R. Wissert, P. Steuer, S. Schopp, R. Thomann and R. Mulhaupt, Macromol. Mater. Eng., 2010, 295, 1107 CrossRef CAS.
- V. H. Pham, T. T. Dang, S. H. Hur, E. J. Kim and J. S. Chung, ACS Appl. Mater. Interfaces, 2012, 4, 2630 CAS.
- S. Vadukumpully, J. Paul, N. Mahanta and S. Valiyaveettil, Carbon, 2011, 49, 198–205 CrossRef CAS.
- S. Wang, M. Tambraparni, J. Qiu, J. Tipton and D. Dean, Macromolecules, 2009, 42, 5251–5255 CrossRef CAS.
- J. Liu, W. Yang, L. Tao, D. Li, C. Boyer and T. P. Davis, J. Polym. Sci., Part A: Polym. Chem., 2010, 48, 425 CrossRef CAS.
- H. J. Salavagione, A. M. Dez-Pascual, E. Lazaro, S. Vera and M. A. Gomez-Fatou, J. Mater. Chem. A, 2014, 2, 14289 CAS.
- T. Ramanathan, A. A. Abdala, S. Stankovich, D. A. Dikin, M. Herrera-Alonso, R. D. Piner, D. H. Adamson, H. C. Schniepp, X. Chen, R. S. Ruoff, S. T. Nguyen, I. A. Aksay, R. K. Prud'Homme and L. C. Brinson, Nat. Nanotechnol., 2008, 3, 327 CrossRef CAS PubMed.
- I.-H. Kim and Y. G. Jeong, J. Polym. Sci., Part B: Polym. Phys., 2010, 48, 850 CrossRef CAS.
- P. Seurer, R. Wissert, R. Thomann and R. Mulhaupt, Macromol. Rapid Commun., 2009, 30, 316 CrossRef PubMed.
- X. Wang, L. Song, H. Yang, H. Lu and Y. Hu, Ind. Eng. Chem. Res., 2011, 50, 5376 CrossRef CAS.
- Y. Guo, C. Bao, L. Song, B. Yuan and Y. Hu, Ind. Eng. Chem. Res., 2011, 50, 7772 CrossRef CAS.
- X. Wang, Y. Hu, L. Song, H. Yang, W. Xing and H. Lu, J. Mater. Chem., 2011, 21, 4222 RSC.
- H. Hu, X. Wang, J. Wang, L. Wan, F. Liu, H. Zheng, R. Chen and C. Xu, Chem. Phys. Lett., 2010, 484, 247 CrossRef CAS.
- T. Kuila, S. Bose, P. Khanra, N. H. Kim, K. Y. Rhee and J. H. Lee, Composites, Part A, 2011, 42, 1856 CrossRef.
- A. Patole, S. P. Patole, H. Kang, J. B. Yoo, T. H. Kim and J. H. Ahn, J. Colloid Interface Sci., 2010, 350, 530 CrossRef CAS PubMed.
- M. M. Gudarzi and F. Sharif, J. Colloid Interface Sci., 2012, 366, 44 CrossRef CAS PubMed.
- K. Song, Y. Zhang, J. Meng, E. C. Green, N. Tajaddod, H. Li and M. L. Minus, Materials, 2013, 6, 2543 CrossRef CAS.
- P. Król, in Linear Polyurethanes: Synthesis Methods, Chemical Structures, Properties and Application, CRC Press, Boca Raton, FL, 1st edn, 2008, ch. 5, pp. 24–29 Search PubMed.
- G. Diaconu, M. Micusik, A. Bonnefond, M. Paulis and J. R. Leiza, Macromolecules, 2009, 42, 3316 CrossRef CAS.
Footnote |
| † Electronic supplementary information (ESI) available: FTIR spectra of nanocomposites are compared with that of polyurethane prepolymer. See DOI: 10.1039/c4ra16406g |
| This journal is © The Royal Society of Chemistry 2015 |