
Palladium-catalyzed synthesis of aldehydes from aryl halides and tert-butyl isocyanide using formate salts as hydride donors†
Abstract
An efficient one-pot palladium-catalyzed hydroformylation of aryl halides to produce aromatic aldehydes has been achieved, employing tert-butyl isocyanide as a C1 resource and formate salt as a hydride donor without any additional bases. Characterized by its mild reaction conditions, easy operation and lower toxicity, this reaction can tolerate a wide array of functional groups with moderate to excellent yields.
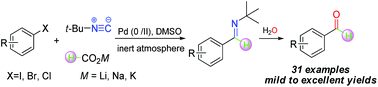
 Please wait while we load your content...
Please wait while we load your content...

