
Lipase-catalyzed green synthesis of enantiopure atenolol†
Bharat Prasad Dwivedee,
Saptarshi Ghosh,
Jayeeta Bhaumik,
Linga Banoth and
Uttam Chand Banerjee*
Department of Pharmaceutical Technology (Biotechnology), National Institute of Pharmaceutical Education and Research, Sector 67, S. A. S. Nagar-160062, Punjab, India. E-mail: ucbanerjee@niper.ac.in; Fax: +91-172-2214692; Tel: +91-172-2214682-85 ext. 2142
First published on 16th January 2015
Abstract
A new green route is proposed for the synthesis of enantiopure atenolol (a β1-blocker). An enzymatic kinetic resolution approach was used to synthesize the enantiopure intermediates (R)- and (S)-2-(4-(3-chloro-2-hydroxypropoxy)phenyl)acetamide from the corresponding racemic alcohol. Of the commercially available lipases screened, Candida antarctica lipase-A (CLEA) showed maximum enantioselectivity in the transesterification of the racemic alcohol using vinyl acetate as the acyl donor. The reactions afforded the (S)-alcohol along with the (R)-acetate, with 48.9% conversion (E = 210, eeP = 96.9% and eeS = 91.1%). Various reaction parameters were optimized in order to achieve maximum enantioselectivity. N-alkylation of the (S)-alcohol with isopropylamine afforded the (S)-atenolol, and the (R)-acetate was chemically hydrolyzed to the corresponding alcohol and further converted to the (R)-atenolol via N-alkylation of the (R)-alcohol with isopropylamine. The use of ionic liquids, to solve the solubility related problems of the drug intermediates, made this process greener and more efficient compared to the previously reported methods.
1. Introduction
β-Blockers are an important class of drugs, which are used in the treatment of several cardiac, migraine and anxiety disorders.1–5 However, many commercially available β-blockers are still used in their racemic form. Chirality is a key factor to determine the efficacy of β-blockers.6–8 The synthesis of a single enantiomeric form of β-blockers is in increasing demand in the pharmaceutical industry.8 Atenolol is a highly polar β1-selective blocker, which cannot penetrate the blood–brain barrier9. Atenolol is used in the treatment of high blood pressure, hypertension, angina, acute myocardial infarction, tachycardia, and the symptoms of alcohol withdrawal.10 Similar to the other β-blockers, atenolol is used as a racemic mixture, despite the fact that its maximum β1-blocking activity is attributed to the (S)-enantiomer.11,12 Atenolol is an aryloxypropanol amine derivative bearing one chiral centre. In general, the β-blocking activity of the S-enantiomer of an aryloxypropanol amine derivative is 50–500 times higher than that of the (R)-enantiomer. Only few reports are available for the enantiopure synthesis of atenolol. Various catalysts used in the synthesis include sulfated tungstate, CsF, Sm(OTf)3, Jacobsen’s catalyst [(R,R)-(salen Co(III)OAc)], a bimetallic chiral cobaltsalen-type complex and microwave dielectric heating, (strategies 1–6). This demonstrates that only few methods are available for the enantiopure synthesis of this important class of drug molecules. Most of the reported chemical reagents are costly and not environmentally friendly, and require harsh conditions, and also the yield and enantiomeric excess are poor. The reported biocatalytic approach has the main drawback of using an additional chemical modification step to solve the solubility problem (strategy 7) Scheme 1.13–20 | ||
| Scheme 1 Strategies for the synthesis of atenolol. | ||
The increasing popularity of biocatalysis in the field of asymmetric synthesis and the recent work from our group involving the lipase-mediated kinetic resolution for the synthesis of enciprazine, esmolol and bunitrolol led our attention to this problem.21–23 Here, we propose a new strategy (strategy 8) which addresses the issues related to the reported biocatalytic process.17 This strategy involves kinetic resolution with a lipase converting the racemic halohydrin to its enantiopure form followed by amination to produce enantiopure atenolol.
2. Results and discussion
Green chemistry tools for designing new synthetic routes are the need of the hour. The integration of biocatalysis will open new dimensions for the scientific society.24–27 Here, we propose a new chemo-enzymatic route for the (R)- and (S)-atenolol synthesis based on an enzymatic kinetic resolution approach (Scheme 2). The synthesis of the intermediates in an aqueous medium and enzymatic kinetic resolution in an ionic liquid make this process greener compared to the other reported methods. | ||
| Scheme 2 New chemo-enzymatic route for (R)- and (S)-atenolol. | ||
2.1 Synthesis of (RS)-2-(4-(3-chloro-2-hydroxypropoxy)phenyl)acetamide (RS-4)
The starting racemic epoxide (RS)-3 was synthesized through the reaction of 1 with (RS)-2 in the presence of NaOH in water at 4 °C (yield, 90%). The reaction of (RS)-3 with acetyl chloride in methanol and water allowed the formation of the desired substrate (RS)-4 for the lipase-catalyzed kinetic resolution (Scheme 3). | ||
| Scheme 3 Synthesis of (RS)-4. | ||
Standard samples of (R)- and (S)-4 and the corresponding O-acylated derivatives (R)- and (S)-5 are necessary for the optimization of the experimental conditions for the enzymatic kinetic resolution.
2.2 Synthesis of enantiopure 2-(4-(3-chloro-2-hydroxypropoxy)phenyl)acetamide (4)
Using the modified method, compound 1 was treated with (R)-2 and (S)-2, to eventually afford standard (S)-4 and (R)-4, respectively.28 The alkylation using (R)-2 resulted in compound (R)-3 (88% yield) and (S)-2 afforded (S)-3 (85% yield) (Scheme 4). The possible mechanism for the formation of (S)-4 from (R)-2 via (R)-3 can be a nucleophilic epoxide ring opening at the least substituted carbon atom of the epoxide ring [SN2(concerted) mechanism] followed by the nucleophilic displacement of the chlorine atom. Similarly, the (R)-4 derivative can be synthesized from (S)-2 through the (S)-3 intermediate. | ||
| Scheme 4 Mechanism of the (S)-4 formation from (R)-2. | ||
2.3 Synthesis of (RS)/(R)/(S)-1-(4-(2-amino-2-oxoethyl)phenoxy)-3-chloropropan-2-yl acetate (5)
The reaction of (RS)-4 with Ac2O at room temperature under neat conditions in the presence of pyridine (2 mmol) produced (RS)-5 in 94% yield. Acetylation of (R)-4 and (S)-4 using a similar procedure resulted in the formation of (R)-5 and (S)-5 (94% and 93% yield, respectively). The optical purity of both products was determined using chiral HPLC (see ESI sections 6–8†).3. Lipase-catalyzed kinetic resolution of (RS)-4
The complete solubility of a substrate is a crucial factor for any enzymatic reaction. The solubility of substrate (RS)-4 was tested in different organic solvents. However, it was found to be insoluble/partially soluble in most of the solvents (Table 1). The substrate was soluble in isopropyl alcohol (IPA), methanol and DMSO; nevertheless, these were not suitable for the lipase reaction.| S. no. | Solvent | Solubility |
|---|---|---|
| a + Solubility of substrate in organic solvent. − Insolubility of substrate in organic solvent. | ||
| 1. | Dichloromethane | − |
| 2. | Acetonitrile | − |
| 3. | Ethyl acetate | − |
| 4. | Acetone | − |
| 5. | Toluene | − |
| 6. | Isopropyl alcohol | + |
| 7. | Water | − |
| 8. | Methanol | + |
| 9. | Diisopropylether | − |
| 10. | Tetrahydrofuran | Partial |
| 11. | Cyclohexane | − |
| 12. | Benzene | − |
| 13. | Vinyl acetate | Partial |
| 14. | Heptane | − |
| 15. | Dimethylsulfoxide | + |
| 16. | Isooctane | − |
Hence, the solubility of (RS)-4 in ionic liquids was investigated. Three different ionic liquids were investigated for the solubility of (RS)-4 (Table 2).
| S. no. | Ionic liquid | Solubility |
|---|---|---|
| a + Solubility of substrate in ionic liquid. − Insolubility of substrate in ionic liquid. | ||
| 1. | [BMIM]BF4 | − |
| 2. | [EMIM]BF4 | + |
| 3. | [BMIM]PF6 | − |
It was found that (RS)-2-(4-(3-chloro-2-hydroxypropoxy)phenyl)acetamide was only soluble in [EMIM]BF4. Hence, [EMIM]BF4 was selected for further reaction with the lipase. For the kinetic resolution, (RS)-4 (20 mmol, 1 eq.) was dissolved in the ionic liquid ([EMIM]BF4, 200 μL) and further reacted with various lipase preparations (commercially available) in the presence of vinyl acetate as the acyl donor (100 μL) in toluene (900 μL) for 30 h. The reaction mixture was then extracted with IPA and the solvent was evaporated under vacuum. Samples for HPLC analysis were prepared using IPA solvent.
3.1 Screening of lipases
A screening of enzymes is the first step to achieve successful kinetic resolution for any transesterification reaction. Lipases from commercially available sources [immobilized lipase in sol–gel–Ak from Pseudomonas cepacia (PCL), immobilized lipozyme from Mucor meihei (MML), lipase acrylic resin from Candida antarctica (CAL), Candida antarctica lipase-A (CAL-A) CLEA, Candida rugosa (CRL L8525), Candida rugosa (CRL L1754), Candida rugosa (CRL 62316), Candida cylindracea (CCL), Aspergillus niger (ANL), porcine pancreas lipase (PPL), lipase AY “Amano”30 (CRL)] were screened for the transesterification of (RS)-4 with vinyl acetate in toluene (Scheme 5).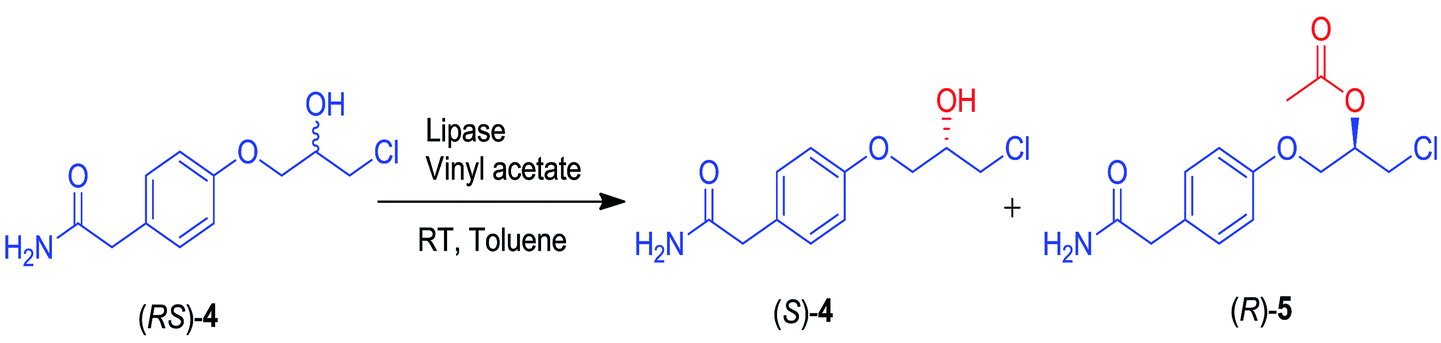 | ||
| Scheme 5 Enzymatic kinetic resolution of (RS)-4. | ||
Amongst all the lipases screened (total 11), Candida antarctica lipase A (CLEA) exhibited the best enantioselectivity for the conversion of (RS)-4 to (R)-4 and (S)-5. Thus, Candida antarctica lipase A (CLEA) was found to be better in terms of conversion and enantioselectivity (Table 3). See ESI section 9† for selective HPLC chromatograms of lipase screening.
| S. no. | Lipase | Time (h) | Cb (%) | eeSc (%) | eePd (%) | Ee | Selectivity |
|---|---|---|---|---|---|---|---|
a Conditions: (RS)-4 (20 mM) in toluene (1 mL) was treated with vinyl acetate (5.4 mmol) at 30 °C in the presence of the CAL-A (CLEA) lipase, 15 mg enzyme preparation with a 5.72 U mg−1 activity was used (total activity was 85.7 IU mL−1 in the reaction mixture).b Conversions were calculated from the enantiomeric excesses (ee) of (S)-4 (substrate S) and (R)-5 (product P) using the formula: Conversion (C) = eeS/(eeS + eeP).c Enantiomeric excess of (S)-4 determined via HPLC analysis (Daicel Chiralcel OD-H column, 83![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) :17, hexane:2-propanol, 1 mL min−1 flow rate at 254 nm).d Enantiomeric excess of (R)-5 determined via HPLC analysis (Daicel Chiralcel OD-H column, 83:17, hexane:2-propanol, 1 mL min−1 flow rate at 254 nm).e E value calculated using the formula E = [ln(1 − C(1 + eeP))]/[ln(1 − C(1 − eeP)].34 :17, hexane:2-propanol, 1 mL min−1 flow rate at 254 nm).d Enantiomeric excess of (R)-5 determined via HPLC analysis (Daicel Chiralcel OD-H column, 83:17, hexane:2-propanol, 1 mL min−1 flow rate at 254 nm).e E value calculated using the formula E = [ln(1 − C(1 + eeP))]/[ln(1 − C(1 − eeP)].34 |
|||||||
| 1. | CCL | 30 | 27 | 21 | 58 | 4.7 | S |
| 2. | ANL | 30 | 16 | 4.3 | 23 | 1.7 | RS |
| 3. | MML | 30 | 15 | 2.4 | 14 | 1.4 | RS |
| 4. | CAL | 30 | 0.23 | 0.21 | 90 | 16 | R |
| 5. | CAL-A (CLEA) | 30 | 46.4 | 83.5 | 96.4 | 142 | S |
| 6. | PCL | 30 | 23 | 19 | 64 | 5.4 | RS |
| 7. | CRL62316 | 30 | 26 | 24 | 70 | 7.1 | S |
| 8. | CRL 90860 | 30 | 15 | 13 | 71 | 7.0 | RS |
| 9. | CRL L1754 | 30 | 15 | 14 | 79 | 9.6 | R |
| 10. | AMANO | 30 | 16 | 14 | 75 | 8.2 | S |
| 11. | PPL | 30 | 0 | 0 | 0 | 0 | RS |
3.2 Optimization of process parameters for the kinetic resolution
In order to maximize the conversion and enantiomeric excess of the product, the effects of different reaction parameters on the activity and enantioselectivity of the lipase were optimised.| S. no. | Solvent | logP |
Cb (%) | eeSc (%) | eePd (%) | Ee |
|---|---|---|---|---|---|---|
| a Conditions: (RS)-4 (20 mM) in toluene (1 mL) was treated with vinyl acetate (5.4 mmol) at 30 °C in the presence of the CAL-A (CLEA) lipase, 15 mg enzyme preparation with a 5.72 U mg−1 activity was used (total activity was 85.7 IU mL−1 in the reaction mixture).b Conversions were calculated from the enantiomeric excesses (ee) of (S)-4 (substrate S) and (R)-5 (product P) using the formula: Conversion (C) = eeS/(eeS + eeP).c Enantiomeric excess of (S)-4 determined by HPLC analysis (Daicel Chiralcel OD-H column, 83:17, hexane:2-propanol, 1 mL min−1 flow rate at 254 nm).d Enantiomeric excess of (R)-5 determined by HPLC analysis (Daicel Chiralcel OD-H column, 83:17, hexane:2-propanol, 1 mL min−1 flow rate at 254 nm).e E value calculated using the formula E = [ln(1 − C(1 + eeP)]/[ln(1 − C(1 − eeP)].34 |
||||||
| 1. | Acetonitrile | −0.3 | 0 | 0 | 0 | 0 |
| 2. | 1,4-Dioxane | −1.1 | 37 | 44 | 75 | 11 |
| 3. | Diethylether | 0.85 | 26 | 18 | 51 | 3.6 |
| 4. | Dichloromethane | 1.3 | 25 | 19 | 57 | 4.4 |
| 5. | Tertiary butyl methyl ether | 1.4 | 15 | 13 | 72 | 6.8 |
| 6. | Benzene | 2.0 | 31 | 34 | 75 | 9.8 |
| 7. | Toluene | 2.5 | 48.5 | 91.1 | 96.9 | 210 |
The solvent effect on the activity and enantioselectivity of CAL-A (CLEA) for the kinetic resolution of (RS)-4 was studied using vinyl acetate as the acyl donor at 30 °C. It was observed that both the reaction rate and enantioselectivity were largely affected by the solvent employed (Table 4). For CAL-A (CLEA), toluene was found to offer a maximum enantioselectivity and enantiomeric excess of the substrate and product as compared to the other solvents (Fig. 1). See ESI section 10† for selective HPLC chromatograms of solvent screening.
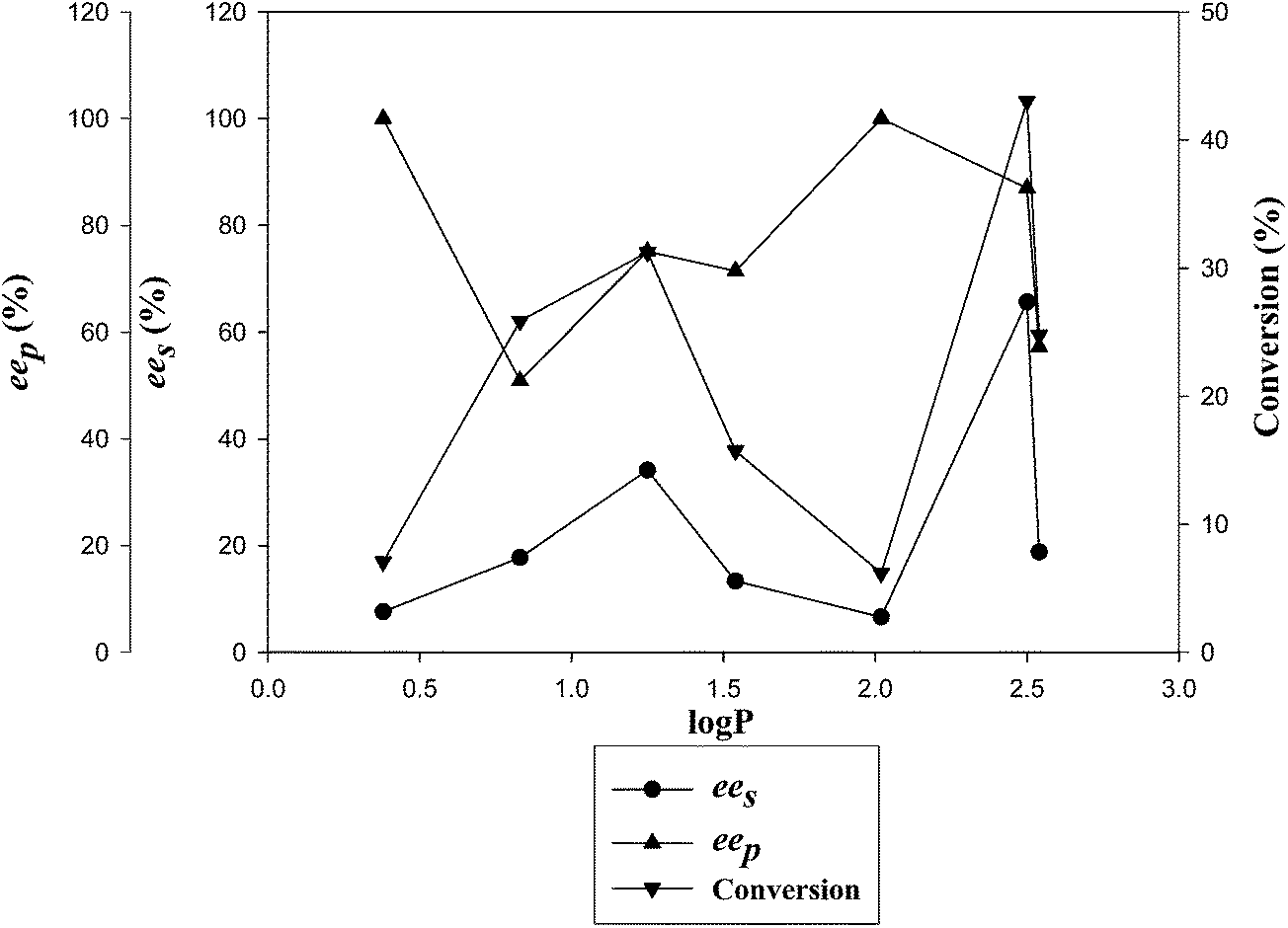 | ||
| Fig. 1 Effect of solvent on CAL-A (CLEA)-catalyzed transesterification of (RS)-4. | ||
000 × g for 5 minutes to remove the enzyme preparation. The conversion and enantiomeric excess were determined using chiral HPLC. It was observed that the conversion and enantiomeric excess of the substrate increased with the reaction time. Maximum conversion (C = 48%, eeS = 91%, eeP = 97%) was achieved after 18 h of reaction time and thereafter no significant changes in the conversion rate and enantiomeric excess were observed (Fig. 2). Thus, 18 h was chosen as the optimum time to perform further studies (see ESI no. 11† for HPLC chromatogram for the optimized time period).
 | ||
| Fig. 2 Course of reaction of CAL-A (CLEA)-catalyzed transesterification of (RS)-4 in toluene. | ||
| S. no. | Acyl donorsa | Cb (%) | eePc (%) | eeSd (%) | Ee |
|---|---|---|---|---|---|
| a Conditions: (RS)-4 (20 mM) in toluene (1 mL) was treated with vinyl acetate (5.4 mmol) at 30 °C in the presence of the CAL-A (CLEA) lipase, 15 mg enzyme preparation with a 5.72 U mg−1 activity was used (total activity was 85.7 IU mL−1 in the reaction mixture).b Conversions were calculated from the enantiomeric excesses (ee) of (S)-4 (substrate S) and (R)-5 (product P) using the formula: Conversion (C) = eeS/(eeS + eeP).c Enantiomeric excess of (S)-4 determined using HPLC analysis (Daicel Chiralcel OD-H column, 83:17, hexane:2-propanol, 1 mL min−1 flow rate at 254 nm).d Enantiomeric excess of (R)-5 determined using HPLC analysis (Daicel Chiralcel OD-H column, 83:17, hexane:2-propanol, 1 mL min−1 flow rate at 254 nm).e E value calculated using the formula E = [ln(1 − C(1 + eeP)]/[ln(1 − C(1 − eeP)].34 |
|||||
| 1. | Benzyl acetate | 0 | 0 | 0 | 0 |
| 2. | Ethyl acetate | 0 | 0 | 0 | 0 |
| 3. | Isopropenyl acetate | 10 | 96 | 11 | 57 |
| 4. | Vinyl acetate | 48.4 | 97.0 | 91.1 | 210 |
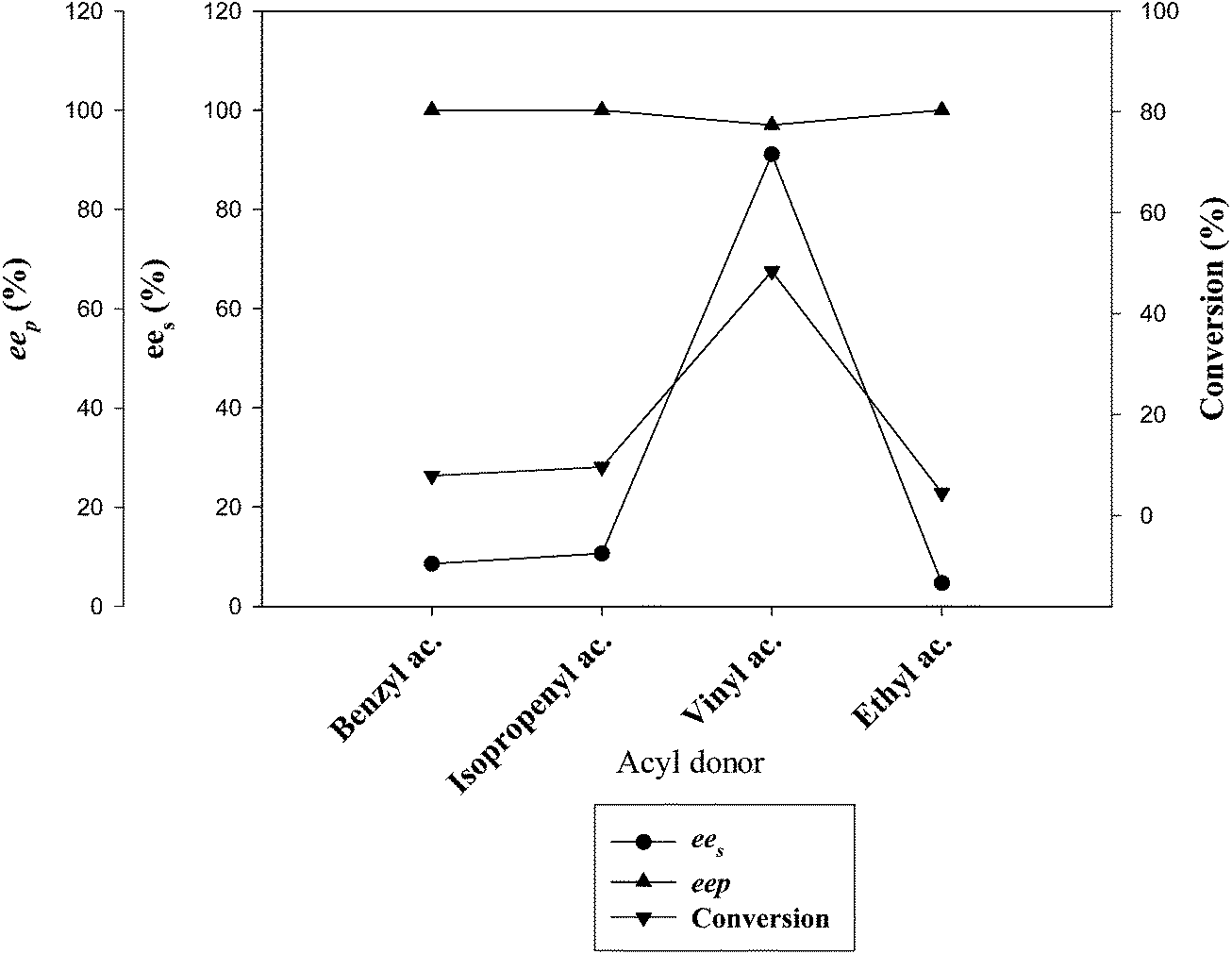 | ||
| Fig. 3 Effect of various acyl donors on the kinetic resolution of (RS)-4. | ||
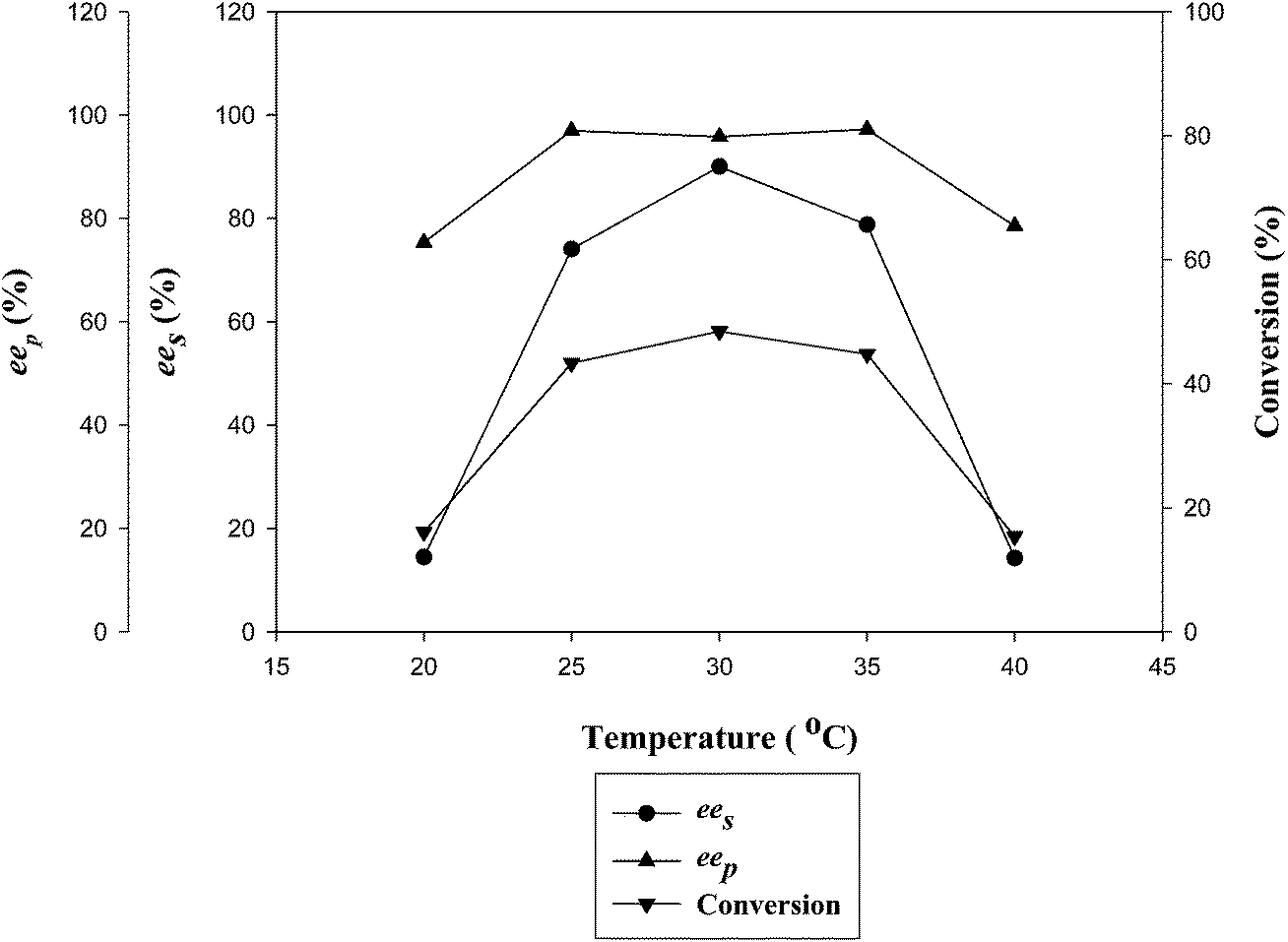 | ||
| Fig. 4 Effect of reaction temperature on the kinetic resolution of (RS)-4. | ||
 | ||
| Fig. 5 Effect of enzyme concentration on the CAL-A (CLEA)-catalyzed transesterification of (RS)-4 in toluene. | ||
 | ||
| Fig. 6 Effect of substrate concentration on the kinetic resolution of (RS)-4. | ||
3.3 Synthesis of (R)-4 through deacylation of (R)-5
The deacetylation of (R)-5 in aqueous K2CO3 at room temperature resulted in (R)-4 (Scheme 6). | ||
| Scheme 6 Synthesis of (R)-4. | ||
3.4 Synthesis of (S)-atenolol
(S)-4, which was obtained via enzymatic kinetic resolution of (RS)-4, was treated with isopropylamine overnight to obtain the (S)-atenolol 6 (Scheme 7).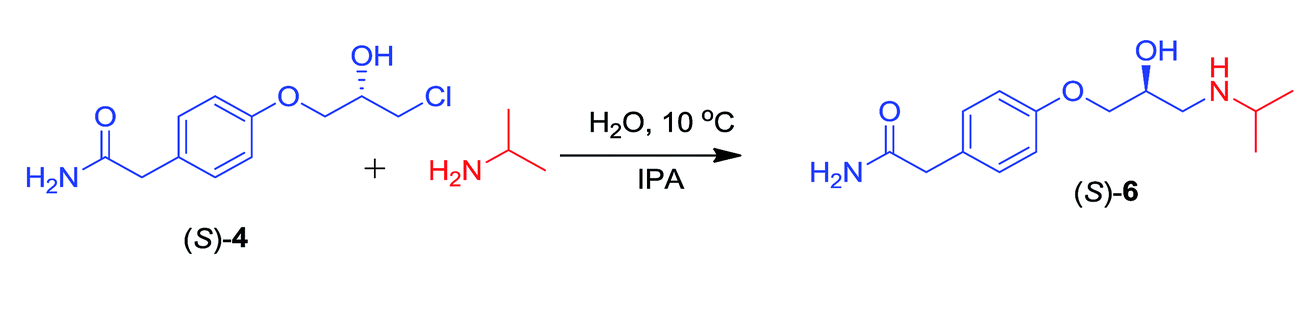 | ||
| Scheme 7 Synthesis of the (S)-atenolol (S-6). | ||
3.5 Synthesis of (R)-atenolol
(R)-4, obtained from the deacetylation of (R)-5, was treated with isopropylamine and the reaction was carried out overnight to afford the (R)-atenolol 6 (Scheme 8). | ||
| Scheme 8 Synthesis of the (R)-atenolol (R-6). | ||
4. Experimental section
4.1 General experimental details
:2-propanol (83:17) was taken as the mobile phase (flow rate: 1 mL min−1, 25 °C).4.2 Synthesis of 2-(4-(3-chloro-2-hydroxypropoxy)phenyl)acetamide (4)
An aqueous solution of (RS)-epichlorohydrin (20 mmol, 1.5 eq.) was cooled to 4 °C. A solution of 4-hydroxyphenyl acetamide (1) and tetra butyl ammonium bromide (0.001–2% by weight of 4-hydroxyphenyl acetamide) in aq. sodium hydroxide (20 mmol, 1 eq.) was then added to the above-mentioned mixture. The resulting solution was then stirred at 4 °C until the TLC (DCM/methanol, 19:1) showed completion based on the starting material (50 h). The resulting precipitate was filtered, washed with water and dried at 60 °C to give a mixture of glycidylether and chlorohydrin, which was characterized using GCMS. The reaction mixture was directly used in the next step without further purification.
Acetyl chloride was added to the above-mentioned reaction mixture in a methanol–water solvent system. The reaction was continued at room temperature for 3 h and the completion was confirmed via TLC (DCM/methanol, 19:1) analysis. Evaporation of methanol resulted in the formation of a precipitate, which was filtered, washed with water and dried at 60 °C to afford the following chlorohydrin derivative:
(RS)-2-(4-(3-chloro-2-hydroxypropoxy)phenyl)acetamide (RS-4), white solid (92% yield);
1H NMR (400 MHz; DMSO-d6; Me4Si) δ 3.27 (s, 2H, CH2Cl), 3.69 (m, 2H, COCH2), 3.99 (d, J = 4.8 Hz, 2H, OCH2), 4.02 (d, J = 5.2 Hz, 1H, CHOH), 5.6 (d, J = 5.3 Hz, 1H, COH), 6.85 (d, J = 8.4 Hz, 2H, ArH), 7.16 (d, J = 8.6 Hz, 2H, ArH), 7.43 (br s, 2H, NH2).
The product was then subjected to chiral HPLC analysis using a chiral OD-H column. The two enantiomers were eluted at tS = 41.5 min and tR = 44.3 min (83:17, hexane:2-propanol).
4.3 Synthesis of (RS)-1-(4-(2-amino-2-oxoethyl)phenoxy)-3-chloropropan-2-ylacetate (5)
(RS)-5 was chemically synthesized through the treatment of (RS)-4 (0.5 mmol, 1 eq.) with Ac2O (1 mL) in pyridine (0.5 mmol, 1 eq.) at 30 °C for 2 h. After consumption of (RS)-4, ice water (50 mL) was added into the reaction mixture and the pH was adjusted to 3 by adding 3 M HCl. The resulting product was extracted with ethyl acetate and washed with brine solution. The organic layer was then separated and concentrated under vacuum to afford (RS)-5 as a colourless liquid (96% yield).(RS)-1-(4-(2-amino-2-oxoethyl)phenoxy)-3-chloropropan-2-ylacetate (RS)-5, colourless liquid (96% yield).
1H NMR (400 MHz; CDCl3; Me4Si) δ 2.11 (m, 3H, COCH3), 3.51 (s, 2H, CH2Cl), 3.82 (m, 2H, COCH2), 4.17 (m, 2H, OCH2), 5.33 (m, 1H, CHOAc), 5.55 (br s, 1H, NH), 6.02 (br s, 1H, NH), 6.90 (m, 2H, ArH), 7.20 (m, 2H, ArH).
(RS)-5 was then subjected to chiral HPLC analysis using a chiral OD-H column. The two enantiomers were eluted at tR = 29.1 min and tS = 36.4 min (83:17, hexane:2-propanol).
4.4 Enantioselective transesterification of (RS)-4
A mixture of (RS)-4 (20 mmol) in 100 μL [EMIM]BF4, 200 μL vinyl acetate and 900 μL toluene was placed in a 5 mL conical flask. Lipases from different sources [Candida antarctica lipase-A (CAL-A), Candida rugosa 90860, Candida rugosa 62316, Candida rugosa L-1754, Candida cylindracea, Aspergillus niger, porcine pancreas and AY “Amano”30, immobilized lipase in sol–gel–Ak from Pseudomonas cepacia, immobilized lipozyme from Mucor miehei, lipase acrylic resin from Candida antarctica, Candida antarctica lipase-A (CAL-A) CLEA] were used to carry out the reaction. All enzyme preparations were individually put into separate 5 mL conical flasks containing the reaction mixture. The flasks were capped and placed in a shaker which was maintained at 30 °C (200 rpm). The samples were extracted from the reaction mixture using isopropyl alcohol (IPA); the conversion and enantiomeric excesses of the substrates and the products of the reactions were monitored using HPLC.
4.5 Optimization of transesterification reaction
The effect of different organic solvents such as acetonitrile, 1,4-dioxane, tert-butyl methyl ether, diisopropylether, diethylether, dichloromethane, benzene, heptane, isooctane and toluene on the transesterification of (RS)-4 was observed. The optimum time was determined by carrying out the reaction and collecting the samples at various time intervals. Enzyme preparations of different activity (e.g. 85.8, 172, 257, 343, 514 IU mL−1) with respect to their concentrations (15, 30, 45, 60 and 90 mg mL−1) were used with a fixed substrate concentration (20 mmol). Different substrate concentrations (e.g. 10, 20, 30, 40, and 50 mmol) were used to find the optimum concentration in the reaction mixture. The samples were analyzed by determining the enantioselectivity in the transesterification reaction.4.6 General method for the deacylation of (RS)/(R)/(S)-5
A solution of K2CO3 (2 mmol) in distilled water (1 mL) was added to a solution of compound 5 (1 mmol) in methanol (5 mL) and the resultant reaction mixture was allowed to stir for 2 h at room temperature. Upon completion, the reaction mixture was extracted with dichloromethane and washed with water. The organic extracts were combined, dried over Na2SO4 and concentrated under vacuum to obtain the crude which was purified via silica gel column chromatography (100–200 mesh) to afford the corresponding alcohol.(RS)-2-(4-(3-chloro-2-hydroxypropoxy)phenyl)acetamide (RS-4), white solid (95% yield).
1H NMR (400 MHz; DMSO-d6; Me4Si) δ 3.27 (s, 2H, CH2Cl), 3.69 (m, 2H, COCH2), 3.99 (d, J = 4.8 Hz, 2H, OCH2), 4.02 (d, J = 5.2 Hz, 1H, CHOH), 5.6 (d, J = 5.3 Hz, 1H, COH), 6.85 (d, J = 8.4 Hz, 2H, ArH), 7.16 (d, J = 8.6 Hz, 2H, ArH), 7.43 (br s, 2H, NH2).
(R)-4: white solid, (92% yield). The product was then subjected to chiral HPLC analysis using a Chiralcel OD-H column, and the two enantiomers were eluted at tS = 40.5 min and tR = 45.3 min (hexane:2-propanol, 83:17, flow rate 1 mL min−1) with peak areas of 1.0% and 99%, respectively (98% ee).
(S)-4: white solid, (93.5% yield). The product was then subjected to chiral HPLC analysis using a Chiralcel OD-H column, and the two enantiomers were eluted at tS = 40.5 min and tR = 45.3 min (hexane:2-propanol, 83:17, 1 mL min−1) with peak areas of 99% and 1.0%, respectively (98% ee).
4.7. General method for the synthesis of (RS)/(S)/(R)-6
A mixture of isopropylamine (5 mL) and water (1 mL) was cooled to 10 °C, (RS)/S-4 (1 mmol) was added and the reaction was stirred for 12 h at 10 °C. Upon completion of the reaction, excess isopropylamine was removed via evaporation and the resulting residue was treated with water. The resulting suspension was acidified with 5 N HCl to adjust the pH to 2.0 and the resulting solution was filtered and washed with water. The filtrate was basified up to pH 11.0 by adding 2 N aq. NaOH and the resulting precipitate was filtered and washed with water and dried to give the corresponding atenolol.2-{4-[2-Hydroxy-3-(propan-2-ylamino)propoxy]phenyl} acetamide (RS-6), white solid (95% yield).
1H NMR (400 MHz; DMSO-d6; Me4Si) δ 0.97 (m, 6H, CH3CHCH3), 2.55 (d, J = 6.0 Hz, 1H, CHCH3), 2.68 (m, 2H, NHCH2), 3.28 (s, 2H, COCH2), 3.83 (d, J = 6.0 Hz, 2H, OCH2), 3.91 (d, J = 4.8 Hz, 1H, CHOH), 5.06 (br s, 1H, COH), 6.85 (d, J = 8.6 Hz, 2H, ArH), 7.15 (d, J = 8.6 Hz, 2H, ArH), 7.47 (br s, 2H, NH2).
(R)/(S)-6: white solid (93 and 94% yield, respectively). The characterization data (1H NMR) were consistent with those of the chemically synthesized RS-6.
5. Conclusions
Among various commercial lipases screened, CAL-A (CLEA) showed a prominent enantioselectivity for the transesterification of (RS)-2-(4-(3-chloro-2-hydroxypropoxy) phenyl)acetamide and afforded the key intermediates (R)-1-(4-(2-amino-2-oxoethyl) phenoxy)-3-chloropropan-2-ylacetate and S-2-(4-(3-chloro-2-hydroxypropoxy)phenyl)acetamide with good conversion and enantiomeric excess. The solubility of the intermediates in an ionic liquid (EMIMBF4) resolved the solubility issues associated with the racemic intermediate (RS)-2-(4-(3-chloro-2-hydroxypropoxy)phenyl)acetamide in the lipase-catalyzed reaction, which avoids additional chemical modification steps for solubilization. The enzymatic shift towards the synthesis of enantiopure atenolol is an excellent example of green synthesis. The efficient chemoenzymatic synthesis of the enantiomerically pure cardiovascular drug atenolol was developed with an improved overall yield.Acknowledgements
This work was supported by the National Institute of Pharmaceutical Education and Research, Council of Scientific and Industrial Research and Department of Biotechnology, Govt. of India, New Delhi. B. P. Dwivedee would like to thank the Department of Biotechnology, Govt. of India, for financial support.References
- P. Larochelle and S. W. Tobe, Can. J. Cardiol., 2014, 30, S16–S22 CrossRef PubMed
.
- H. L. Kennedy, Am. J. Med., 2001, 110, 25–65 Search PubMed
- R. Mehvar and D. R. Brocks, J. Pharm. Pharm. Sci., 2001, 4, 185–200 CAS
- Prescriber, 2010, 21, 50–54 Search PubMed.
- W. H. Frishman, Circulation, 2003, 107, 117–119 CrossRef PubMed
- S. W. Tobe, Can. J. Cardiol., 2014, 30, S1–S2 CrossRef PubMed
- M. Maciejewski, K. Połtorak and J. E. Kaminska, J. Mol. Catal. B: Enzym., 2010, 62, 248–256 CrossRef CAS PubMed
- R. N. Patel, Biomolecules, 2013, 3, 741–777 CrossRef PubMed
- P. Agon, P. Goethals, D. V. Haver and J. M. Kaufman, J. Pharm. Pharmacol., 2011, 43, 597–600 CrossRef PubMed
- B. Carlberg, O. Samuelsson and L. H. Lindholm, Lancet, 2004, 364, 1684–1689 CrossRef CAS
- J. Agustian, A. H. Kamaruddin and S. Bhatia, Proc. Biochem., 2010, 45, 1587–1604 CrossRef CAS PubMed
- L. M. Kuyper and N. A. Khan, Can. J. Cardiol., 2014, 30, S47–S53 CrossRef PubMed
- S. P. Pathare and K. G. Akamanchi, Tetrahedron Lett., 2013, 54, 6455–6459 CrossRef CAS PubMed
- D. S. Bose and V. A. Narsaiah, Bioorg. Med. Chem., 2005, 13, 627–630 CrossRef CAS PubMed
- S. M. Jang and T. S. Shieh., USP, Patent no: 5290598, 1994
- S. R. Mehta, B. M. Bhawal, V. H. Deshpande and M. K. Gurjar. USP Patent no: 6982 349, 2004
- H. S. Bevinakatti and A. A. Banerji, J. Org. Chem., 1992, 57, 6003–6005 CrossRef CAS
- J. S. Yadav, A. R. Reddy, A. V. Narsaiah and B. V. S. Reddy, J. Mol. Catal. A: Chem., 2007, 261, 207–212 CrossRef CAS PubMed
- R. B. Kawthekar, W. Bi and G. Kim, Appl. Organomet. Chem., 2008, 22, 583–591 CrossRef CAS
- R. B. Kawthekar and G. Kim, Helv. Chim. Acta, 2008, 91, 317–332 CrossRef CAS
- L. Banoth, K. N. Thete and U. C. Banerjee, Tetrahedron: Asymmetry, 2012, 23, 1272–1278 CrossRef CAS PubMed
- L. Banoth, B. Chandarrao, B. Pujala, A. K. Chakraborti and U. C. Banerjee, Synthesis, 2014, 46, 479–488 Search PubMed
- L. Banoth and U. C. Banerjee, Arabian J. Chem., 2014 DOI:10.1016/j.arabjc.2014.03.011
- K. Alfonsi and M. Stefaniak, Green Chem., 2008, 10, 31–36 RSC
- S. D. Roughley and A. M. Jordan, J. Med. Chem., 2011, 54, 3451–3479 CrossRef CAS PubMed
- C. M. Clouthier and J. N. Pelletier, Chem. Soc. Rev., 2012, 41, 1585–1605 RSC
- L. Banoth, M. Singh, A. Tekewe and U. C. Banerjee, Biocatal. Biotransform., 2009, 27, 263–270 CrossRef CAS
- P. W. Erhardt, C. M. Woo, W. G. Anderson and R. J. Gorczynski, J. Med. Chem., 1982, 25, 1408–1412 CrossRef CAS
-
(a) Y. L. Khmelnitsky and J. O. Rich, Curr. Opin. Chem. Biol., 1999, 3, 47–53 CrossRef CAS
- J. S. Dordick, Enzyme Microb. Technol., 1989, 11, 194–211 CrossRef CAS
- R. Chênevert, N. Pelchat and P. Morin, Tetrahedron: Asymmetry, 2009, 20, 1191–1196 CrossRef PubMed
- M. Paravidino and U. Hanefeld, Green Chem., 2011, 13, 2651–2657 RSC
- G. Egri, E. Baitz-Gács and L. Poppe, Tetrahedron: Asymmetry, 1996, 7, 1437–1448 CrossRef CAS
- A. J. J. Straathof and J. A. Jongejan, Enzyme Microb. Technol., 1997, 21, 559–571 CrossRef CAS
Footnote |
| † Electronic supplementary information (ESI) available: HPLC chromatograms (lipase screening, solvent, time, temperature, enzyme and substrate concentration optimization), HRMS and NMR spectra of selected compounds. See DOI: 10.1039/c4ra16365f |
| This journal is © The Royal Society of Chemistry 2015 |