
Organocatalysis in polysiloxane gels: a magnetic-stir-bar encapsulated catalyst system prepared by thiol–ene photo-click immobilization†
Hong Yang*,
Ming Xu,
Ling-Xiang Guo,
Hao-Fan Ji,
Jun-Yu Wang,
Bao-Ping Lin*,
Xue-Qin Zhang and
Ying Sun
School of Chemistry and Chemical Engineering, Jiangsu Province Hi-Tech Key Laboratory for Bio-medical Research, Southeast University, Nanjing 211189, China. E-mail: yangh@seu.edu.cn; lbp@seu.edu.cn; Fax: +86 25 52091096; Tel: +86 25 52091096
First published on 22nd December 2014
Abstract
This manuscript presents a facile thiol–ene photo-click chemistry method to prepare magnetic stir bar-encapsulated polysiloxane-based organocatalyst gels under benign conditions, and develops a Stir Bar-Encapsulated Catalysis (SBEC) technique. Through thiol–ene addition chemistry, we graft olefin-terminated organocatalysts (i.e. MacMillan catalyst, proline catalyst, and N-heterocyclic carbene catalyst) onto poly[3-mercaptopropylmethylsiloxane], which is further photo-crosslinked to coat the embedded magnetic stir bar. The prepared magnetic stir bar-encapsulated polysiloxane-based organocatalyst gels can be put into reaction flasks to perform stirring and catalysis functions at the same time. The most important benefit of SBEC technique is to infinitely simplify the catalyst/product separation procedure by using a simple stir-bar-retriever, even without any precipitation/filtration steps. The catalytic performances of three different organocatalyst gels applied in asymmetric Diels–Alder reaction, asymmetric aldol reaction and benzoin condensation reaction respectively are also examined herein.
Introduction
Heterogeneous catalysts, prepared by immobilizing catalysts on surfaces of inorganic materials or functionalized organic polymers,1–3 offer several engineering advantages such as easy separation, high stability and facile catalyst recycling, thus play an important role in “Green Chemistry” processes.4–6 Among various support media, cross-linked polysiloxane gels are very attractive carrier materials and have acquired global interest over the last decade for their wealth of advantageous properties such as good chemical and thermal stabilities, superhydrophobicity, highly flexible Si–O–Si bonds, as well as excellent permeability which allows organic molecules to go through siloxane matrixes with very fast diffusion velocities.7To support catalysts on polysiloxane gels, conventional noncovalent immobilization methods8 are to occlude catalysts such as Grubbs' catalysts,9,10 BINAP-Ru,11,12 Salen-Mn13 and DuPHOS-Rh,14 into polydimethylsiloxane (PDMS) films or slabs. Although this strategy is very convenient and efficient, catalyst leaching even in aqueous solution is an inevitable and serious problem.14 In order to overcome this defect, covalent immobilization method provides another solution by chemically linking the catalysts onto the polysiloxane matrixes. Previously reported protocols always relied on a platinum-catalyzed hydrosilylation reaction of polymethylhydrosiloxane (PMHS) and olefin-terminated catalysts or ligands to build polysiloxane-based catalysts.15–17 However, this approach also has several obvious disadvantages: (1) hydrosilylation reaction efficiency is quite variable and unpredictable so that the catalyst grafting ratio is beyond control; (2) noble metal, platinum is very expensive and meanwhile is difficult to be removed from the products, which might complicate the following catalytic applications; (3) the preparation procedure usually requires long reaction time, plenty of solvents and a high reaction temperature.
Herein, we present a facile thiol–ene photo-click chemistry method18–20 to prepare polysiloxane-gel-based organocatalysts under benign conditions. As shown in Fig. 1, we use instead of PMHS, poly[3-mercaptopropylmethylsiloxane] (PMMS)21,22 which bears one mercapto group in every monomer unit, and graft olefin-terminated organocatalysts (MacMillan catalyst C1, proline catalyst C2, and N-heterocyclic carbene (NHC) catalyst C3) onto PMMS chain. Meanwhile, by mixing the above systems with a photo-initiator (2,2-dimethoxy-2-phenylacetophenone, DMPA) and a variety of olefin-functional crosslinkers, a series of organocatalyst-immobilized polysiloxane gels can be synthesized by UV-initiated thiol–ene click chemistry. Compared with traditional hydrosilylation procedure, this thiol–ene photo-click protocol, as a greener and cleaner approach, has an almost 100% reaction conversion; uses cheap photo-initiators as catalysts, which are much easier to be removed; and requires very mild reaction conditions such as minute-scale reaction time, solvent-less environment-friendly process and ambient temperature, etc.
 | ||
| Fig. 1 Preparation methods and molecular structures of polysiloxane-gel-based organocatalysts herein this manuscript. | ||
Furthermore, inspired by Stir Bar-Sorptive Extraction (SBSE) technique,23 we use organocatalyst-immobilized PMMS gel instead of PDMS, to coat magnetic stir bar, and develop a Stir Bar-Encapsulated Catalysis (SBEC) technique. As shown in Fig. 2, a plastic vial containing a magnetic stir bar and the mixture of PMMS, organocatalyst C1, photoinitiator DMPA and crosslinker L3, was UV illuminated for 20 minutes (Fig. 2A–C) to form a cross-linked gel (Fig. 2D). After breaking up the plastic vial, the prepared magnetic stir bar-encapsulated polysiloxane-based organocatalyst gel (Fig. 2E) could be put into a reaction flask to perform stirring and catalysis functions at the same time (Fig. 2F). The intrinsic motivation and the most important benefit of this approach are to infinitely simplify the catalyst/product separation procedure to using a simple stir-bar-retriever (Fig. 2G), even without any precipitation/filtration steps.
 | ||
| Fig. 2 Preparation protocol of magnetic stir bar-encapsulated polysiloxane-based organocatalyst gels: (A) a magnetic stir bar and plastic pipette-head vials. (B) The plastic vial was filled with a magnetic stir bar and the mixture of PMMS, organocatalyst C1, DMPA and crosslinker L3. The oily mixture was UV-illuminated (C) and became a crosslinked gel (D), which was cut out off the vial (E). (F) The obtained organocatalyst gel was performing both stirring and catalysis functions (S1.avi†). (G) A stir-bar-retriever was used to separate the catalyst from products (S2.avi†). | ||
Experimental section
Materials and instrumentation
Poly[3-mercaptopropylmethylsiloxane] (PMMS, SMS-992, Mw 4000–7000, 95 cst) was purchased from Gelest Inc. Poly(ethylene glycol)diacrylate (average Mn ∼700) was purchased from Aldrich Inc. 2,2-Dimethoxy-2-phenylacetophenone (DMPA), (s)-phenylalanine methylester hydrochloride, allylamine, trans-4-hydroxy-L-proline, undec-10-enoyl chloride, 4,5-diphenylimidazole and 11-bromo-1-undecene were purchased from Aladdin (Shanghai) Inc. Dichloromethane, toluene and DMF were distilled from CaH2 under argon. THF was distilled from sodium-benzophenone ketyl under argon. Other chemical reagents were used without further purification. All non-aqueous reactions were conducted in oven-dried glassware, under a dry nitrogen atmosphere. All flash chromatography was performed using Macherey-Nagel MN Kieselgel 60 (0.063–1.2 mm).All 1H NMR spectra were obtained using a Bruker HW500 MHz spectrometer (AVANCE AV-500) and recorded in CDCl3 (internal reference 7.26 ppm). The enantiomeric excess (ee) values were analyzed by Waters 1525 High-performance liquid chromatography (HPLC) with chiral columns. A UV lamp (20 mW cm−2, λ = 365 nm; LP-40A; LUYOR Corporation) was used to irradiate the samples to perform the photo-crosslinking reactions.
![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) :5, 10 mL), stir bar-encapsulated polysiloxane-based organocatalyst gel PMMS-g-C1L3 (estimated as 50 mol%, if all the grafted catalysts could be reached) and CF3COOH (0.29 g, 2.5 mmol) were added. To the above solution freshly distilled cyclopentadiene (1.65 g, 25.0 mmol) were then added. The reaction mixture was stirred at 0 °C for 24 h. The solution was extracted by ethylacetate (3 × 50 mL). The catalyst gel PMMS-g-C1L3 was removed by a stir bar retriever and immerse-washed by CH2Cl2 several times, stored for future uses. The resulting organic layer was washed by brine (2 × 40 mL), dried over MgSO4 and was further concentrated under vacuum to provide a yellow oil. The crude product was further converted into the corresponding alcohol by reduction with an excess NaBH4 in CH3OH at 24 °C for 1 h. The endo/exo ratios were determined by crude NMR, and enantiomeric excess (ee) values were analyzed by chiral HPLC with Daciel Chiralcel OJ-H column (eluent: hexane/isopropanol 7/3; 0.8 mL min−1, λ = 225 nm).:1 petroleum ether/ethylacetate) to give the desired product as a yellow solid. The anti/syn ratios and enantiomeric excess (ee) values were analyzed by chiral HPLC with Daciel Chiralpak AD-H column (eluent: isohexane/isopropanol 9/1; 1.0 mL min−1, λ = 254 nm).:1 petroleum ether/ethylacetate) to give the desired benzoin product (490 mg, yield: 63%) as a white solid. 1H NMR (500 MHz, CDCl3): δ 7.91 (m, 2H), 7.51 (m, 1H), 7.39 (m, 2H), 7.30 (m, 5H), 5.95 (s, 1H).
:5, 10 mL), stir bar-encapsulated polysiloxane-based organocatalyst gel PMMS-g-C1L3 (estimated as 50 mol%, if all the grafted catalysts could be reached) and CF3COOH (0.29 g, 2.5 mmol) were added. To the above solution freshly distilled cyclopentadiene (1.65 g, 25.0 mmol) were then added. The reaction mixture was stirred at 0 °C for 24 h. The solution was extracted by ethylacetate (3 × 50 mL). The catalyst gel PMMS-g-C1L3 was removed by a stir bar retriever and immerse-washed by CH2Cl2 several times, stored for future uses. The resulting organic layer was washed by brine (2 × 40 mL), dried over MgSO4 and was further concentrated under vacuum to provide a yellow oil. The crude product was further converted into the corresponding alcohol by reduction with an excess NaBH4 in CH3OH at 24 °C for 1 h. The endo/exo ratios were determined by crude NMR, and enantiomeric excess (ee) values were analyzed by chiral HPLC with Daciel Chiralcel OJ-H column (eluent: hexane/isopropanol 7/3; 0.8 mL min−1, λ = 225 nm).:1 petroleum ether/ethylacetate) to give the desired product as a yellow solid. The anti/syn ratios and enantiomeric excess (ee) values were analyzed by chiral HPLC with Daciel Chiralpak AD-H column (eluent: isohexane/isopropanol 9/1; 1.0 mL min−1, λ = 254 nm).:1 petroleum ether/ethylacetate) to give the desired benzoin product (490 mg, yield: 63%) as a white solid. 1H NMR (500 MHz, CDCl3): δ 7.91 (m, 2H), 7.51 (m, 1H), 7.39 (m, 2H), 7.30 (m, 5H), 5.95 (s, 1H).Results and discussion
The synthetic protocols of olefin-terminated organocatalyst monomers including MacMillan catalyst C1, proline catalyst C2, and NHC catalyst C3 are shown in Scheme 1. The ester-amide exchange of (S)-phenylalanine methyl ester hydrochloride with allyl amine, followed by condensation reaction with acetone gave the imidazolinone catalyst C1.24 Proline catalyst C2 was prepared by a selective O-acylation of trans-4-hydroxy-L-proline 3 in trifluoroacetic acid.25 Starting from 4,5-diphenylimidazole 4, NHC catalyst C3 was synthesized in two steps by alkylation with 11-bromo-1-undecene and further quaterisation treatment with iodomethane.26 The detailed experimental procedures and 1H NMR spectra are listed in the ESI.† | ||
| Scheme 1 Syntheses of olefin-terminated MacMillan catalyst, proline catalyst, and NHC catalyst. | ||
For preparing magnetic stir bar-encapsulated polysiloxane-based organocatalyst gels, efficient crosslinkage based on the design of crosslinker and crosslinking ratio plays a crucial role in building a stable polymeric network. Based on our previous experiments, commercial PMMS are short oligomers with an estimated degree of polymerization (D.P.) around 30.27,28 Thus, in order to form a stable cross-linked PMMS gel, the molar percentage of the crosslinking sites should be at least higher than 7–8 mol% and herein we set 15 mol% as a constant crosslinking ratio for all the experiments. Three crosslinkers, triallyl cyanurate (L1, TAC), 1,6-hexanediol diacrylate (L2), poly(ethylene glycol)diacrylate (L3, average Mn ∼700) were tested in the experiments respectively. In comparison, although all three crosslinkers could be successfully used to synthesize polysiloxane gels, the gels containing a much longer and flexible crosslinker, poly(ethylene glycol)diacrylate are more elastic and stable than other brittle gels prepared by triallyl cyanurate or 1,6-hexanediol diacrylate crosslinkers.
As shown in Fig. 2, before UV illumination, we first dissolved an organocatalyst into a small amount of methylene chloride which was then mixed with PMMS, photoinitiator and crosslinker to form an oily liquid. The mixture was then poured into a vial containing a magnetic stir bar. Herein, we chose a soft plastic container (PE pipette head, Fig. 2A) in stead of glass vials, because compared with scissor-cut plastic pieces, shattered glass would easily damage the prepared gels in the last step. After UV illumination, the oily liquid became a crosslinked gel (Fig. 2D), which was cut out of the plastic vial and immersed in dry methylene chloride several times to wash out unreacted small molecules. The desired magnetic stir bar-encapsulated polysiloxane-based organocatalyst gel was prepared. However, our prototype manufacturing system has two technique problems: (1) magnetic stir bars are randomly embedded in PMMS gels and we can not precisely arrange the locations and postures of stir bars placed in the gels. Thus, the prepared organocatalyst gels will have physically vulnerable points where the stir bars touch on the walls of plastic container, and this imperfectness results in a moderate stirring effect (see the stirring movie, SI1.avi†). (2) The organocatalyst gels are partially crosslinked and would be better to be stored in organic solvents to maintain elasticity. For example, we have tried to remove all the solvent from the gels via vacuum, which unfortunately caused the spontaneous fission of gels.
Although some flaws exist in our prototype products at present, future industrial manufacturing can be expected to realize technique improvements. Herein, we prepared three magnetic-stir-bar-encapsulated organocatalyst gels, PMMS-g-C1L3, PMMS-g-C2L3 and PMMS-g-C3L3, which were used in catalyzing asymmetric Diels–Alder reaction, asymmetric aldol reaction and benzoin condensation reaction respectively.
The imidazolidinone compound developed by MacMillan,29 might be the most famous organocatalyst which has been widely used in a variety of organocatalytic processes and has been unsurprisingly immobilized on different polymeric supports.24,25,30–34 Polysiloxane gel catalyst, PMMS-g-C1L3 bearing MacMillan imidazolidinone was applied to promote a classical asymmetric Diels–Alder reaction of cyclopentadiene and cinnamic aldehyde.
As shown in Table 1, roughly 50%:50% mixture of endo and exo cycloadducts (determined by 1H NMR analysis of crude products) were isolated in all the experimental trials. To convert the grafted imidazolidinone C1 to the catalytically active intermediate, an equimolar amount of a Bronsted acid is required to protonate the supported organocatalyst. Traditionally, HBF4 has been proven to be a very efficient acid in this reaction system,24 however in our case, this choice provided negative results (entry 1), possibly due to the strong lewis acidity and F− ion of HBF4 which might be able to destroy C–S–C and Si–O bonds. The alternative use of trifluoroacetic acid in acetonitrile/water (95/5) solvent provided an optimal 88% yield with moderate endo (96%) and exo (78%) ee values (entry 3). The recovered PMMS-g-C1L3 gel was recycled five times to test the catalytic performances. As can be seen from the reported data (entry 4–8), the conversion efficiency and catalyst stereoselectivity were maintained at around 70% reaction yield and 77% ee, although slightly lower than the first trial's result. Nonetheless, PMMS-g-C1L3 gel can be conveniently employed to catalyze asymmetric Diels–Alder cycloadditions.
|
||||||
|---|---|---|---|---|---|---|
| Entrya | Recycle number | Acid | Solvent | Yieldb (%) | Exo/endoc (%) | Exo ee (endo ee)d (%) |
| a Reactions were carried out using cinnamic aldehyde (1 equiv.) and cyclopentadiene (5 equiv.) at 0 °C for 24 h.b Isolated yield.c Determined by crude NMR.d Determined by chiral HPLC. | ||||||
| 1 | 0 | HBF4 | CH3CN/H2O (95/5) | 0 | — | — |
| 2 | 0 | TFA | MeOH/H2O (95/5) | 52 | 51/49 | 66 (83) |
| 3 | 0 | TFA | CH3CN/H2O (95/5) | 88 | 51/49 | 78 (96) |
| 4 | 1 | TFA | CH3CN/H2O (95/5) | 73 | 55/45 | 77 (77) |
| 5 | 2 | TFA | CH3CN/H2O (95/5) | 66 | 53/47 | 78 (79) |
| 6 | 3 | TFA | CH3CN/H2O (95/5) | 74 | 51/49 | 74 (77) |
| 7 | 4 | TFA | CH3CN/H2O (95/5) | 72 | 51/49 | 73 (72) |
| 8 | 5 | TFA | CH3CN/H2O (95/5) | 64 | 54/46 | 70 (79) |

Polymer-supported L-proline represents another very important class of organocatalysts for C–C bond constructions such as asymmetric aldol reaction.25,35–46 Following literature protocols, we tested the catalytic performance of polysiloxane gel PMMS-g-C2L3 applied in a classical enantioselective aldol reaction of 4-nitrobenzaldehyde and cyclohexanone. As illustrated in Table 2, solvent plays a crucial role in enantioselective property. The reaction carried out in methanol/H2O (1/1, v/v) system provided moderate yields and low ee (34–38%), while using pure water solution resulted in high conversion (>80%) and high ee values (96–99%). Unlike traditional homogeneous reactions which would have a significant decrease in both stereo- and enantioselectivity along with raising reaction temperature,47–50 our PMMS-g-C2L3 catalyst gel slightly favors higher temperature possibly due to the hydrophobicity of polysiloxanes expelling water from catalytic centers to stabilize the transition state of forming enamine species by excluding competitive hydrogen bonding with water. This phenomena is consistent with Monteiro's observation.46
|
||||||
|---|---|---|---|---|---|---|
| Entrya | Recycle number | Solvent | Temperature (°C) | Yieldc (%) | Anti/synd (%) | Anti eee (%) |
| a Reactions were carried out using 4-nitrobenzaldehyde (1 equiv.) and cyclohexanone (7 equiv.) for 48 h.b Catalyst: PMMS-g-C2L2.c Isolated yield.d Determined by chiral HPLC.e Determined by chiral HPLC. | ||||||
| 1 | 0 | MeOH/H2O (1/1) | 25 | 68 | 89/11 | 38 |
| 2 | 0 | MeOH/H2O (1/1) | 50 | 65 | 92/8 | 34 |
| 3 | 0 | H2O | 25 | 80 | 88/12 | 90 |
| 4b | 0 | H2O | 50 | 82 | 88/12 | 96 |
| 5 | 0 | H2O | 50 | 85 | 90/10 | 99 |
| 6 | 1 | H2O | 50 | 87 | 86/14 | 91 |
| 7 | 2 | H2O | 50 | 76 | 90/10 | 72 |
| 8 | 3 | H2O | 50 | 75 | 81/19 | 60 |
| 9 | 4 | H2O | 50 | 69 | 85/15 | 24 |
| 10 | 5 | H2O | 50 | 73 | 56/44 | 22 |

The recovered PMMS-g-C2L3 gel was reused five times to test the recyclability of catalyzing the asymmetric aldol reaction. As shown in Table 2, the first two runs provided satisfying reaction yields and high ee values (entry 5–6), however the enantioselectivity decreased dramatically starting from the third recycle (entry 7–10). Besides the lack of exploration in optimal reaction conditions, one possible reason might be that since the recovered PMMS-g-C2L3 gel was always kept in solvents to avoid gel fission, some leftover chemicals might “poison” or racemize the grafted L-proline catalyst.
Incorporating imidazolium salts into the polymer backbones or side chains has been proven to be an efficient way to develop recyclable polymeric NHC catalysts.26,51–58 Inspired from Cowley's work,26 we designed and synthesized an imidazolium monomer C3, and grafted it onto crosslinked PMMS gels. With PMMS-g-C3L3 catalyst in hand, we examined its ability of catalyzing a typical benzoin condensation reaction.
As shown in Table 3, the solvent effects were first investigated and we found that DMSO could provide a moderate reaction yield as well as a small amount of benzil product due to the mildly oxidative reaction environment. However, poor recyclability with lower yields (entry 4–8) was observed after the first run, which might arise from the catalyst regeneration step. To regenerate the imidazolium salt from reactive carbene intermediate, a solution of 4.0 M HCl in 1,4-dioxane was used to immerse-wash the recovered PMMS-g-C3L3 gel. Due to the hydrophobicity of polysiloxanes, only the externally grafted imidazolium monomer C3 could be possibly regenerated, which might cause the obvious loss of catalytic performances.
|
||||
|---|---|---|---|---|
| Entrya | Recycle number | Solvent | Yieldb of benzoin product (%) | Yieldb of benzil product (%) |
| a Reactions were carried out using dry DMSO and DBU (15 mol%) at r.t. under N2 for 48 h.b Isolated yield. | ||||
| 1 | 0 | H2O | Trace | 0 |
| 2 | 0 | DMF | 58 | 7 |
| 3 | 0 | DMSO | 63 | 5 |
| 4 | 1 | DMSO | 54 | 9 |
| 5 | 2 | DMSO | 47 | 7 |
| 6 | 3 | DMSO | 32 | 6 |
| 7 | 4 | DMSO | 35 | 4 |
| 8 | 5 | DMSO | 32 | 5 |
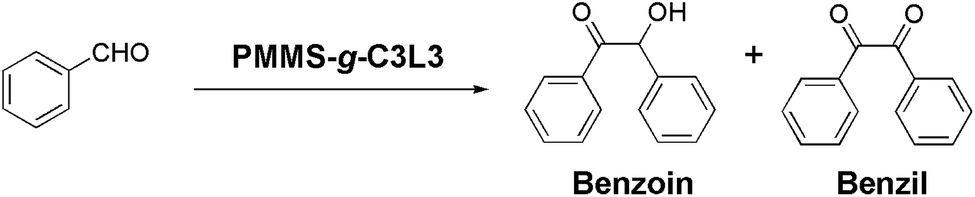
Conclusions
In summary, we describe a facile thiol–ene photo-click chemistry method to prepare magnetic stir bar-encapsulated polysiloxane-based organocatalyst gels under benign conditions. The advantages of this thiol–ene protocol include: green preparation procedure requiring very mild reaction conditions such as minute-scale reaction time, solvent-less environment-friendly process and ambient temperature; almost quantitative grafting and crosslinking conversions; avoidance of using Pt catalysts, etc. However, the disadvantage of this crosslinked PMMS gel system is also obvious: the linkable catalysts are limited to organocatalysts while wide varieties of noble metal catalysts are excluded due to the presence of mercapto groups of PMMS which might poison noble metals.Incorporating magnetic stir bars into crosslinked PMMS gels can provide the corresponding organocatalyst gels an ability to perform stirring and catalysis functions at the same time (SBEC technique). The most important benefit of this technique is to infinitely simplify the catalyst/product separation procedure to using a simple stir-bar-retriever, even without any precipitation/filtration steps. Although our organocatalyst gels are prototype products bearing several technique problems and the catalytic performances are modest, we hope this “proof-of-idea” work would open interesting perspectives and bring some useful information for heterogeneous catalysis community.
Acknowledgements
This research was supported by National Natural Science Foundation of China (Grant no. 21374016). The authors would like to gratefully thank Dr Lin Chen for his help with chiral HPLC experiment measurements.Notes and references
- Handbook of Assymmetric Heterogeneous Catalysis, ed. K. Ding and Y. Uozumi, Viley-VCH, Weinheim, 2008 Search PubMed.
- M. Benaglia, Recoverable and Recyclable Catalysts, ed. M. Benaglia, John Wiley and Sons, 2009 Search PubMed.
- N. Haraguchi and S. Itsuno, Polymeric Chiral Catalyst Design and Chiral Polymer Synthesis, John Wiley and Sons, 2011 Search PubMed.
- Q. H. Fan, Y. M. Li and A. S. C. Chan, Chem. Rev., 2002, 102, 3385–3465 CrossRef CAS PubMed.
- P. McMorn and G. J. Hutchings, Chem. Soc. Rev., 2004, 33, 108–122 RSC.
- M. Heitbaum, F. Glorius and I. Escher, Angew. Chem., Int. Ed., 2006, 45, 4732–4762 CrossRef CAS PubMed.
- J. Chojnowski and M. Cypryk, in Silicon-Containing Polymers-The Science and Technology of Their Synthesis and Applications, ed. R. G. Jones, W. Ando and J. Chojnowski, Springer-Verlag, Dordrecht, The Netherlands, 2000, ch. 1, pp. 3–35 Search PubMed.
- J. M. Fraile, J. I. Garcia and J. A. Mayoral, Chem. Rev., 2009, 109, 360–417 CrossRef CAS PubMed.
- M. T. Mwangi, M. B. Runge and N. B. Bowden, J. Am. Chem. Soc., 2006, 128, 14434–14435 CrossRef CAS PubMed.
- M. B. Runge, M. T. Mwangi and N. D. Bowden, J. Organomet. Chem., 2006, 691, 5287–5288 Search PubMed.
- I. F. J. Vankelecom, D. Tas, R. F. Parton, V. Van de Vyver and P. A. Jacobs, Angew. Chem., Int. Ed., 1996, 35, 1346–1348 CrossRef CAS.
- R. F. Parton, I. F. J. Vankelecom, D. Tas, K. B. M. Janssen, P.-P. Knops-Gerrits and P. A. Jacobs, J. Mol. Catal. A: Chem., 1996, 113, 283–292 CrossRef CAS.
- D. F. C. Guedes, T. C. O. Leod, M. C. A. F. Gotardo, M. A. Schiavon, I. V. P. Yoshida, K. J. Ciuffi and M. D. Assis, Appl. Catal., A, 2005, 296, 120–127 CrossRef CAS PubMed.
- A. Wolfson, S. Janssens, I. Vankelecom, S. Geresh, M. Gottlieb and M. Herskowitz, Chem. Commun., 2002, 388–389 RSC.
- Y. Motoyama, K. Mitsui, T. Ishida and H. Nagashima, J. Am. Chem. Soc., 2005, 127, 13150–13151 CrossRef CAS PubMed.
- Y. Motoyama, M. Abe, K. Kamo, Y. Kosako and H. Nagashima, Chem. Commun., 2008, 5321–5323 RSC.
- Y. Motoyama, K. Kamo and H. Nagashima, Org. Lett., 2009, 11, 1345–1348 CrossRef CAS PubMed.
- M. J. Kade, D. J. Burke and C. J. Hawker, J. Polym. Sci., Part A-1: Polym. Chem., 2010, 48, 743–750 CrossRef CAS.
- G. Franc and A. K. Kakkar, Chem. Soc. Rev., 2010, 39, 1536–1544 RSC.
- C. E. Hoyle and C. N. Bowman, Angew. Chem., Int. Ed., 2010, 49, 1540–1573 CrossRef CAS PubMed.
- L. M. Campos, I. Meinel, R. G. Guino, M. Schierhorn, N. Gupta, G. D. Stucky and C. J. Hawker, Adv. Mater., 2008, 20, 3728–3733 CrossRef CAS.
- L. M. Campos, T. T. Truong, D. E. Shim, M. D. Dimitriou, D. Shir, I. Meinel, J. A. Gerbec, H. T. Hahn, J. A. Rogers and C. J. Hawker, Chem. Mater., 2009, 21, 5319–5326 CrossRef CAS.
- E. Baltussen, P. Sandra, F. David and C. Cramers, J. Microcolumn Sep., 1999, 11, 737–747 CrossRef CAS.
- S. Guizzetti, M. Benaglia and J. S. Siegel, Chem. Commun., 2012, 48, 3188–3190 RSC.
- T. E. Kristensen, K. Vestli, M. G. Jakobsen, F. K. Hansen and T. Hansen, J. Org. Chem., 2010, 75, 1620–1629 CrossRef CAS PubMed.
- A. B. Powell, Y. Suzuki, M. Ueda, C. W. Bielawski and A. H. Cowley, J. Am. Chem. Soc., 2011, 133, 5218–5220 CrossRef CAS PubMed.
- H. Yang, Q. Zhang, B.-P. Lin, G.-D. Fu, X.-Q. Zhang and L.-X. Guo, J. Polym. Sci., Part A-1: Polym. Chem., 2012, 50, 4182–4190 CrossRef CAS.
- H. Yang, M. Liu, Y. Yao, P. Tao, B. Lin, P. Keller, X. Zhang, Y. Sun and L. Guo, Macromolecules, 2013, 46, 3406–3416 CrossRef CAS.
- K. A. Ahrendt, C. J. Borths and D. W. C. MacMillan, J. Am. Chem. Soc., 2000, 122, 4243–4244 CrossRef CAS.
- S. A. Selkala, J. Tois, P. M. Pihko and A. M. P. Koskinen, Adv. Synth. Catal., 2002, 344, 941–945 CrossRef CAS.
- M. Benaglia, G. Celentano, M. Cinquini, A. Puglisi and F. Cozzi, Adv. Synth. Catal., 2002, 344, 149–152 CrossRef CAS.
- A. Puglisi, M. Benaglia, M. Cinquini, F. Cozzi and G. Celentano, Eur. J. Org. Chem., 2004, 567–573 CrossRef CAS.
- Y. Zhang, L. Zhao, S. S. Lee and J. Y. Ying, Adv. Synth. Catal., 2006, 348, 2027–2032 CrossRef CAS.
- N. Haraguchi, Y. Takemura and S. Itsuno, Tetrahedron Lett., 2010, 51, 1205–1208 CrossRef CAS PubMed.
- M. Benaglia, G. Celentano and F. Cozzi, Adv. Synth. Catal., 2001, 343, 171–173 CrossRef CAS.
- D. Font, C. Jimeno and M. A. Pericas, Org. Lett., 2006, 8, 4653–4655 CrossRef CAS PubMed.
- D. Font, S. Sayalero, A. Bastero, C. Jimeno and M. A. Pericas, Org. Lett., 2007, 9, 1943–1946 CrossRef CAS PubMed.
- T. Kehat and M. Portnoy, Chem. Commun., 2007, 2823–2825 RSC.
- M. Gruttadauria, F. Giacalone, A. M. Marculescu, P. Lo Meo, S. Riela and R. Noto, Eur. J. Org. Chem., 2007, 468–4698 Search PubMed.
- M. Benaglia, M. Cinquini, F. Cozzi, A. Puglisi and G. Celentano, Adv. Synth. Catal., 2002, 344, 533–542 CrossRef CAS.
- A. Lu, P. Contanda, J. P. Patterson, D. A. Longbottom and R. K. O'Reilly, Chem. Commun., 2012, 48, 9699–9701 RSC.
- E. Huerta, P. J. M. Stals, E. W. Meijer and A. R. A. Palmans, Angew. Chem., Int. Ed., 2012, 52, 2906–2910 CrossRef PubMed.
- A. Lu, D. Moatsuo, D. A. Longbottom and R. K. O'Reilly, Chem. Sci., 2013, 4, 965–969 RSC.
- C. N. Urbani and M. J. Monteiro, Macromolecules, 2009, 42, 3884–3886 CrossRef CAS.
- K. O. Sebakhy, S. Kessel and M. J. Monteiro, Macromolecules, 2010, 43, 9598–9600 CrossRef CAS.
- H. A. Zayas, A. Lu, D. Valade, F. Amir, Z. Jia, R. K. O'Reilly and M. J. Monteiro, ACS Macro Lett., 2013, 2, 327–331 CrossRef CAS.
- N. Zotova, A. Franzke, A. Armstrong and D. G. Blackmond, J. Am. Chem. Soc., 2007, 129, 15100 CrossRef CAS PubMed.
- N. Zotova, L. J. Broadbelt, A. Armstrong and D. G. Blackmond, Bioorg. Med. Chem. Lett., 2009, 19, 3934–3937 CrossRef CAS PubMed.
- K. N. Rankin, J. W. Gauld and R. J. Boyd, J. Phys. Chem. A, 2002, 106, 5155–5159 CrossRef CAS.
- S. Bahmanyar, K. N. Houk, H. J. Martin and B. List, J. Am. Chem. Soc., 2003, 125, 2475–2479 CrossRef CAS PubMed.
- N. Marion, S. Diez-Gonzalez and S. P. Nolan, Angew. Chem., Int. Ed., 2007, 46, 2988–3000 CrossRef CAS PubMed.
- K. V. S. Ranganath, S. Onitsuka, A. K. Kumar and J. Inanaga, Catal. Sci. Technol., 2013, 3, 2161–2181 CAS.
- A. G. M. Barrett, A. C. Love and L. Tedeschi, Org. Lett., 2004, 6, 3377–3380 CrossRef CAS PubMed.
- J. Pinaud, J. Vignolee, Y. Gnanou and D. Taton, Macromolecules, 2011, 44, 1900–1908 CrossRef CAS.
- B. Karimi and P. F. Akhavan, Chem. Commun., 2009, 3750–3752 RSC.
- K. Thiel, R. Zehbe, J. Roeser, P. Strauch, S. Enthaler and A. Thomas, Polym. Chem., 2013, 4, 1848–1856 RSC.
- P. Coupillaud, J. Pinaud, N. Guidolin, J. Vignolle, M. Fevre, E. Veaudecrenne, D. Mecerreyes and D. Taton, J. Polym. Sci., Part A-1: Polym. Chem., 2013, 51, 4530–4540 CAS.
- U. R. Seo and Y. K. Chung, RSC Adv., 2014, 4, 32371–32374 RSC.
Footnote |
| † Electronic supplementary information (ESI) available. See DOI: 10.1039/c4ra16351f |
| This journal is © The Royal Society of Chemistry 2015 |