DOI:
10.1039/C4RA16347H
(Paper)
RSC Adv., 2015,
5, 10505-10511
Multi-analyte, ratiometric and relay recognition of a 2,5-diphenyl-1,3,4-oxadiazole-based fluorescent sensor through modulating ESIPT†
Received
14th December 2014
, Accepted 5th January 2015
First published on 7th January 2015
Abstract
A new two dipicolyl amine (DPA) dangled 2,5-diphenyl-1,3,4-oxadiazole derivatized sensor L has been designed and synthesized. Sensor L displays excited state intramolecular proton transfer (ESIPT) properties in buffered water solution (HEPES 10 mM, pH = 7.4). L shows highly selective and ratiometric fluorescence responses to Zn2+ in a 1![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) :2 binding ratio. Fluorescence spectra and 1H NMR studies demonstrate that L binds Zn2+ ions through its amide form, which promoted the ESIPT emission enhancement. The in situ generated L–2Zn2+ complex can serve as a relay recognition sensor toward PPi and S2− via further complexation and Zn2+ sequestering processes respectively. Thus, multi-analyte and relay recognition of L through modulating ESIPT has been achieved.
:2 binding ratio. Fluorescence spectra and 1H NMR studies demonstrate that L binds Zn2+ ions through its amide form, which promoted the ESIPT emission enhancement. The in situ generated L–2Zn2+ complex can serve as a relay recognition sensor toward PPi and S2− via further complexation and Zn2+ sequestering processes respectively. Thus, multi-analyte and relay recognition of L through modulating ESIPT has been achieved.
1. Introduction
Zn2+, as the second most abundant transition metal ion in the human body, plays fundamental roles in a variety of biological processes including gene transcription, signal transmission, and mammalian reproduction.1 Early studies proved that disruption of Zn2+ concentration in cells can result in various pathological processes such as Alzheimer's disease, epilepsy and infantile diarrhea.2 Therefore, considerable efforts have been focused on the development of a Zn2+ specific fluorescent sensor due to the high sensitivity and easy operability of fluorescence techniques.3
Pyrophosphate (PPi) detection has attracted immense attention of chemists due to its critical roles in several bioenergetic and metabolic processes.4 PPi is released during the DNA polymerase chain reaction, and higher PPi levels in biological fluids such as blood serum are pertinent to cardiovascular disease and acute renal failure.5 Recent investigations reveal that high synovial fluid PPi is associated with some diseases such as formation of calcium pyrophosphate dehydrate crystals and chondrocalcinosis.6 As a consequence, development of chemosensors for highly selective detection of PPi aroused considerable interest and a great number of PPi selective sensors have been documented.7
On the other hand, sulfide detection has also received considerable attention because of its notorious toxicity. Sulfide has variable utilities in the fields of manufacturing of sulfuric acid, dyes and cosmetics,8 however, exposure to high levels of sulfide is dangerous because over inhalation of sulfide can lead to various physiological and biochemical problems including irritation in mucous membranes, unconsciousness, and respiratory paralysis.9 Consequently, a number of sulfide selective chemosensors have been developed.10 Although sulfide sensing utilizing a metal complex via S2−-induced metal ion displacement approach is a simple and effective method, fluorescent sulfide detection with Zn2+-complexes are still rare.11
Recently, relay recognition, especially the sequential metal ion and anion recognition has received increasing more attention due to the operation simplicity and labor-saving, because the metal ion and anion recognition can accomplished sequentially under the identical conditions.12 Fluorescent sensors with ESIPT property are very attractive due to the two wavelength emissions and large Stokes shift.13 Recent studies demonstrate that it is an effective approach to realize ratiometric recognition through modulating the ESIPT process.11,14
With these ideas in mind, we herein report the design and synthesis of a new oxadiazole derivatized fluorescent sensor L, which is expected to serve as an effective relay recognition sensor for Zn2+ and PPi/S2− through modulating the ESIPT behavior. The designed principles for sensor L are based on the following verities and hypothesis: (1) due to well-known DPA binding units, sensor L may exhibit highly selective and ratiometric response to Zn2+ along with Zn2+ binding induced enhancement of ESIPT;15 (2) the in situ generated L–Zn2+ complex has potential applicability to serve as an effective sensor for some anions such as PPi or S2− through anion direct binding or anion elicit Zn2+ sequestering;10c,14a,16 (3) it is rational to realize ratiometric recognition of Zn2+ and a certain anion through perturbing the ESIPT behavior of the sensor. As we expected, sensor L indeed displays highly selective and ratiometric fluorescence recognition to Zn2+ in water solution, the in situ formed L–Zn2+ complex can serve as a dual fluorescence responses sensor to PPi and S2− through different fluorescence emission properties.
2. Results and discussion
2.1 Fluorescence recognition of L to Zn2+
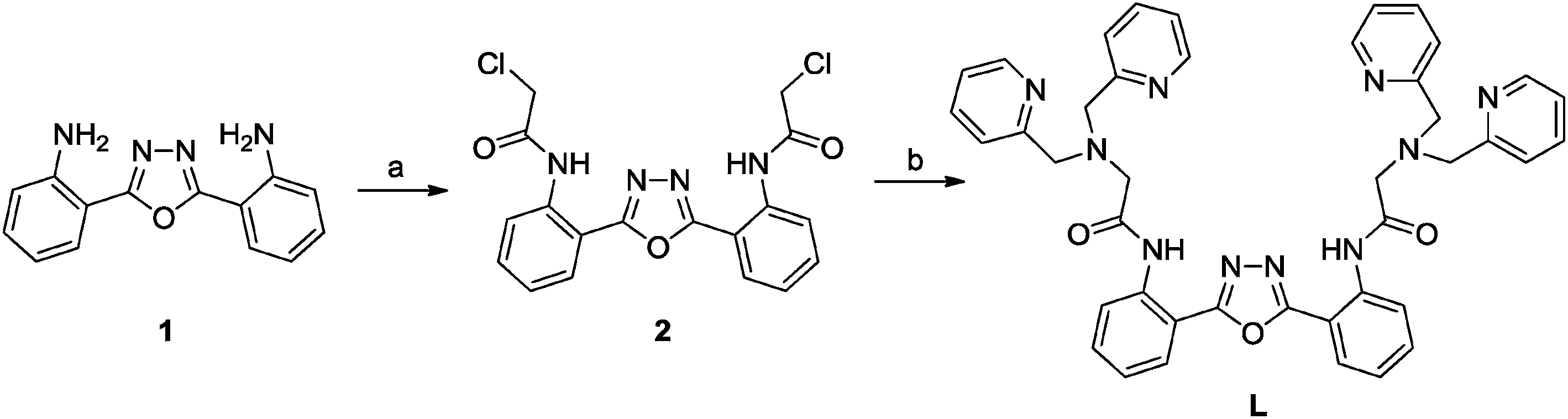
Sensor L was readily prepared as depicted in Scheme 1, and the structure of L was fully characterized by NMR and HRMS spectrometry. In virtue of the good water solubility of L, we could examine its fluorescence properties in HEPES buffered (10 mM, pH = 7.4) solution. Upon excitation at 315 nm, L (10 μM) displays two emission bands appeared at 378 nm and 445 nm, they are attributed to the normal state shorter-wavelength and excited state longer-wavelength emission, respectively. This result indicates that our previous hypothesis that incorporation one more amide group may promote the ESIPT process is true. Subsequently, the metal ion selectivity of L was evaluated (Fig. 1). Upon addition 2.0 equiv. of Zn2+ to L solution, a significant fluorescence intensity enhancement was observed at 445 nm along with a decrease in intensity at 378 nm, indicating that the Zn2+ binding leading to a great enhancement of ESIPT emission of L. However, upon individual addition of other metal ions including Cu2+, Co2+, Hg2+, Ni2+, Ag+, Pb2+, Sr2+, Ba2+, Fe2+, Mn2+, Fe3+, Al3+, Cr3+, Mg2+, K+, Cd2+, and Na+ caused significant fluorescence quenching at 445 nm, particularly Cu2+, Co2+, Hg2+ and Ni2+ induced almost complete fluorescence quenching of L at 445 and 378 nm due to their paramagnetic properties.17 These results demonstrate that the ESIPT process of L was suppressed on interaction with the examined metal ions except Zn2+. Therefore, an excellent selectivity of L to Zn2+ was achieved. In addition, L solution also behaves strong blue fluorescence on addition of Zn2+ under illumination with a portable UV lamp at 254 nm (inset in Fig. 1). The time-dependent fluorescence changes of L solution in the presence of 2.0 equiv. of Zn2+ were then examined. The results reveal that the fluorescence intensity increases with prolonging the stocking time and reaches a plateau after 1.5 h of Zn2+ addition (Fig. S1†). Thus, in the following investigations, the fluorescence spectra were checked after 1.5 h of Zn2+ addition.
 |
| | Scheme 1 Synthesis of sensor L. Reagents and conditions: (a) chloroacetic chloride, AcONa/AcOH; (b) DPA, KI, DIPEA, dry CH3CN, reflux 10 h. | |
 |
| | Fig. 1 Fluorescence spectra changes of L (10 μM) in HEPES buffered (10 mM, pH = 7.4) water solution upon respective addition 2.0 equiv. of different metal ions (λex = 315 nm). Inset: fluorescence color changes of L solution before and after addition of Zn2+ under illumination with a portable UV lamp at 254 nm. | |
Subsequently, the fluorescence titration experiment was conducted to evaluate the sensing behavior of L to Zn2+. As shown in Fig. 2, upon incremental addition of Zn2+ (0 to 2.0 equiv.) to L solution, the emission intensity at 378 nm decreased gradually accompanied with significant enhancement of emission band at 445 nm. This ratiometric fluorescence emission variation terminated when 2.0 equiv. of Zn2+ was employed, indicative of a 1:2 binding stoichiometry of L and Zn2+. Benesi–Hildebrand plot analysis based on 1:2 binding ratio was carried out and resulted in a nice linear straight line (linear coefficient greater than 0.99) (Fig. S2†), further advocates the proposed 1:2 binding ratio of L with Zn2+. The apparent association constant of L and Zn2+ was estimated to be 1.67 × 1010 M−2. Based on the titration data, the detection limit of L to Zn2+ was then calculated. Plotting the normalized fluorescence intensity against log[Zn2+] provided a nice linear relationship (Fig. 3), and the point of this line crossed the ordinate axis is regarded as the detection limit,18 which was found to be 2.77 × 10−6 M.
 |
| | Fig. 2 Fluorescence spectra changes of L (10 μM) in HEPES buffered (10 mM, pH = 7.4) water solution upon incremental addition of Zn2+ (0 to 2.0 equiv.). λex = 315 nm. | |
 |
| | Fig. 3 Changes of normalized fluorescence intensity (F − Fmin)/(Fmax − Fmin) of L (10 μM) against log[Zn2+] in water solution (HEPES 10 mM, pH = 7.4). λex = 315 nm, λem = 445 nm. | |
To further verify the binding ratio of L with Zn2+, Job's plot analysis was performed. The results show that the emission intensity of the tested solution reaches a maximum when the mole ratio of Zn2+ appeared at 0.67, indicating the 1:2 binding stoichiometry of L and Zn2+ (Fig. S3†). A solid evidence for this binding ratio comes from the high-resolution mass spectroscopy (HRMS) analysis of L–Zn2+ solution, the significant peak appeared at m/z 893.1190 is assignable to [L–2H+ + 2Zn2+ + Cl−]+ (Fig. S4†).
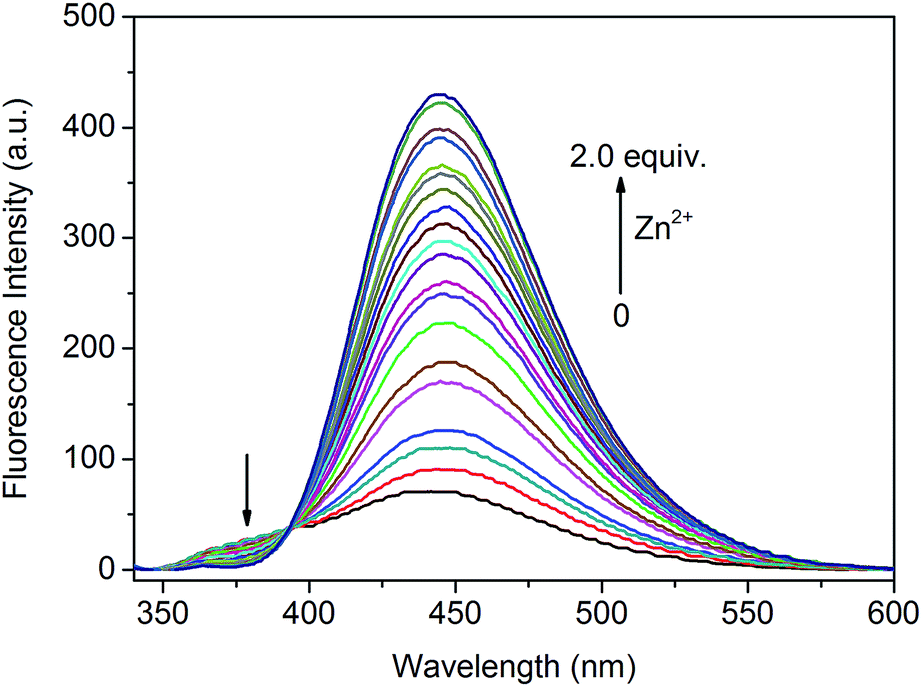
To obtain better insights into the binding behavior of L and Zn2+, 1H NMR spectra of L with and without Zn2+ were compared in DMSO-d6, because the Zn2+ induced enhanced emission band of L in DMSO (λem = 462 nm) is very close to that in buffered water solution (λem = 445 nm) (Fig. S5†), indicating the similar coordination modes of L with Zn2+ in both H2O and DMSO solutions. As shown in Fig. 4, the amide NH proton in free L signaling at 11.50 ppm up-field shifted to 10.82 ppm (Fig. 4b) on addition of Zn2+, suggesting that L binds Zn2+ through an amide form.3d,19 The methylene protons (Ha) adjacent to C![[double bond, length as m-dash]](https://www.rsc.org/images/entities/char_e001.gif) O appeared at 3.44 ppm (Fig. 4a) downfield shifted to 3.86 ppm after binding with Zn2+ (Fig. 4b). Concomitantly, the singlet peak of methylene protons (Hb) in DPA moiety at 3.87 ppm (Fig. 4a) down-field shifted to ca. 4.40 and divided into two nonequivalent groups (Fig. 4b), indicating that the free rotation of sigma bonds in DPA unit of L are restricted after coordination with Zn2+. The proton signal at 8.51 ppm (Fig. 4a) could be tentatively assigned to Hd due to the possible hydrogen bonding between Hd and amide O atom.15 After binding with Zn2+, this signal up-field shifted to ca. 8.0 ppm (Fig. 4b), indicative of the chelation of amide O with Zn2+, which suppressed the hydrogen bonding. Protons neighboring pyridine N atom (Hc) signaling at 8.38 ppm (Fig. 4a) downfield shifted to 8.72 ppm (Fig. 4b) on addition of Zn2+. These results are very similar to those we previously reported sensor OXD for Zn2+ recognition.15 Thus, the 1H NMR investigations demonstrate that L binds with Zn2+ through an amide form. Based on these results, the proposed binding mode of L and Zn2+ is illustrated in Fig. 4.
O appeared at 3.44 ppm (Fig. 4a) downfield shifted to 3.86 ppm after binding with Zn2+ (Fig. 4b). Concomitantly, the singlet peak of methylene protons (Hb) in DPA moiety at 3.87 ppm (Fig. 4a) down-field shifted to ca. 4.40 and divided into two nonequivalent groups (Fig. 4b), indicating that the free rotation of sigma bonds in DPA unit of L are restricted after coordination with Zn2+. The proton signal at 8.51 ppm (Fig. 4a) could be tentatively assigned to Hd due to the possible hydrogen bonding between Hd and amide O atom.15 After binding with Zn2+, this signal up-field shifted to ca. 8.0 ppm (Fig. 4b), indicative of the chelation of amide O with Zn2+, which suppressed the hydrogen bonding. Protons neighboring pyridine N atom (Hc) signaling at 8.38 ppm (Fig. 4a) downfield shifted to 8.72 ppm (Fig. 4b) on addition of Zn2+. These results are very similar to those we previously reported sensor OXD for Zn2+ recognition.15 Thus, the 1H NMR investigations demonstrate that L binds with Zn2+ through an amide form. Based on these results, the proposed binding mode of L and Zn2+ is illustrated in Fig. 4.
 |
| | Fig. 4 1H NMR spectrum of L (DMSO-d6) in the absence (a) and presence (b) of Zn2+. | |
As a good sensor, the anti-interference ability is also important. Thus, the influences of other potential competitive metal ions were assayed. As we previously described, individual addition of other metal ion except Zn2+ produced different extent emission quenching of L at 445 nm. On further addition of Zn2+ to L solution containing other metal ion (except Cu2+), noticeable fluorescence enhancement at 445 nm could be observed (Fig. 5). Even though Co2+ and Hg2+ induced moderate interferences on Zn2+ recognition event, the substantial emission signal enhancements are clear enough to signify the existence of Zn2+. Coexistence of Cu2+ could totally suppress the fluorescence emission of L, indicating that L binds Cu2+ much more tightly than that of Zn2+, which excludes the metal ion exchange. These results demonstrate that the Zn2+ recognition process has a good anti-interference ability to other metal ions except Cu2+.
 |
| | Fig. 5 Fluorescence intensity (at 445 nm) changes of L (10 μM) in water solution (HEPES 10 mM, pH = 7.4) in the presence of various metal ions. The gray bars represent the fluorescence intensity of L solution in the presence of 2.0 equiv. of miscellaneous metal ions; the red bars represent the fluorescence intensity of the above solution upon further addition of 2.0 equiv. of Zn2+. λex = 315 nm. | |
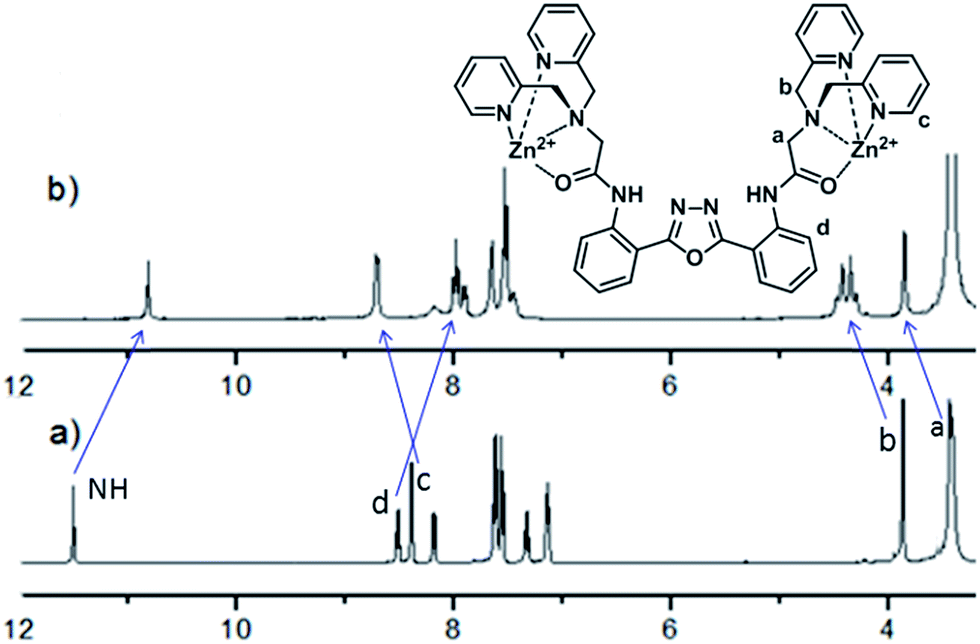
To further confirm the practicability of L for Zn2+ detection, pH effect on fluorescence changes of L (at 445 nm) with and without Zn2+ was then examined (Fig. 6). L solution exhibits weak fluorescence emission within pH range from 2 to 13. In the presence of 2.0 equiv. of Zn2+, the solution displays strong fluorescence emission from pH 7 to 10, suggesting that sensor L is suitable for Zn2+ detection at near neutral pH conditions.
 |
| | Fig. 6 The fluorescence intensity changes (at 445 nm) of L and L–2Zn2+ solution at various pH conditions. λex = 315 nm. | |
2.2 Relay recognition of L–2Zn2+ to S2− and PPi
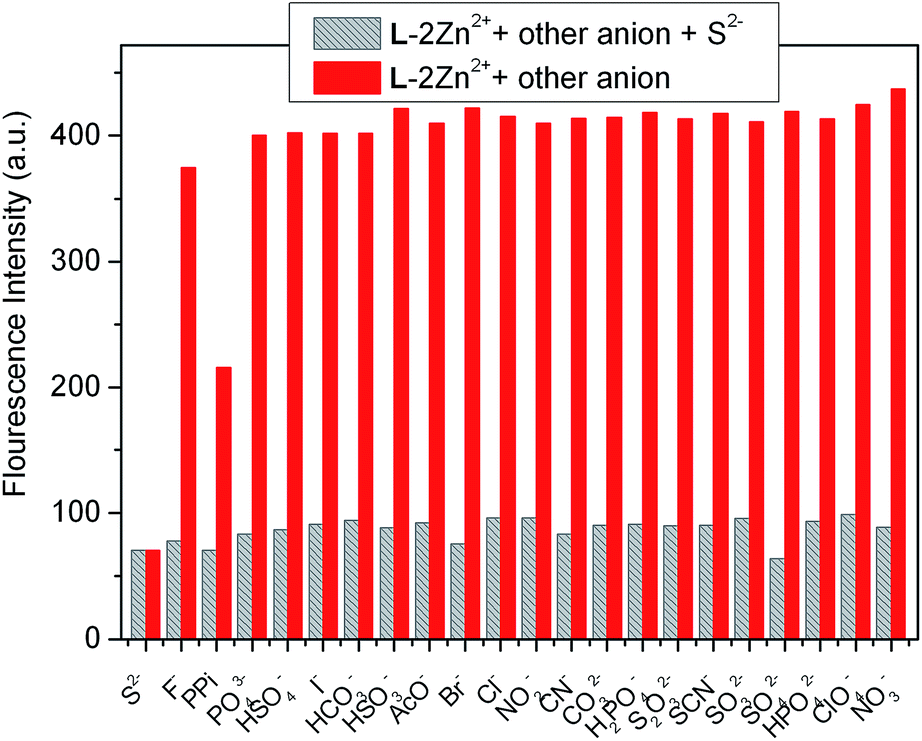
With the relay recognition idea in mind, we then examined the fluorescence response of L–2Zn2+ complex to different anions. L–2Zn2+ solution for anion sensing was in situ prepared by simply mixing 2.0 equiv. of Zn2+ with L. As depicted in Fig. 7, addition of S2− to L–2Zn2+ solution elicited fluorescence band recovery of L, suggesting the S2−-induced Zn2+ ion sequestering event. On addition of PPi, a new emission band at 396 nm with 49 nm blue shift was observed. Other interested anions including F−, Cl−, Br−, I−, SCN−, PO43−, S2O32−, H2PO4−, HPO42−, NO2−, NO3−, AcO−, ClO4−, CN−, C2O42−, SO42−, P2O74−, HSO4−, CO32−, and HCO3−, induced almost negligible fluorescence emission changes of L–2Zn2+ solution. These results demonstrate that L–2Zn2+ complex can selectively detect S2− and PPi through different fluorescence responses.
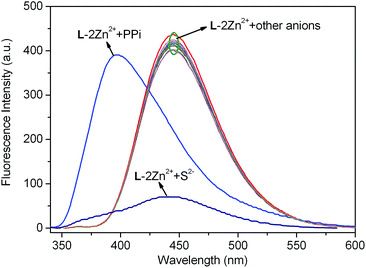 |
| | Fig. 7 Fluorescence spectra changes of the in situ generated L–2Zn2+ solution (10 μM, HEPES 10 mM, pH = 7.4) upon respective addition 5.0 equiv. of different anions. λex = 315 nm. | |
2.2.1 S2− recognition by L–2Zn2+ complex. To evaluate the S2− sensing behavior of L–2Zn2+, fluorescence titration experiments of L–2Zn2+ solution with various amounts of S2− was then conducted. Upon gradual addition of S2− to L–2Zn2+ solution, the fluorescence intensity at 445 nm gradually decreased alone with increase in emission intensity at 378 nm (Fig. 8). When 3.0 equiv. of S2− was added, the emission bands restored to the emission state of L, indicating that the bound Zn2+ in L–2Zn2+ complex was sequestered by S2− and releasing the free L. This S2−-induced Zn2+ sequestering process was further proved by 1H NMR spectra comparison. The 1H NMR spectrum (in DMSO-d6) of L–2Zn2+ in the presence of S2− almost exactly restored to that of free L (Fig. S6†), this result strongly supports the Zn2+ displacement event. According to the titration profile, the detection limit of L–2Zn2+ to S2− was evaluated to be 2.28 × 10−6 M following the aforementioned method (Fig. S7†). Competition experiments were then conducted and the results indicate that the S2− recognition process is hardly influenced by other coexisting anions except PPi (Fig. 9).
 |
| | Fig. 8 Fluorescence spectra changes of the in situ generated L–2Zn2+ solution (10 μM, HEPES 10 mM, pH = 7.4) upon addition of different amount of S2− (0 to 3.0 equiv.). λex = 315 nm. | |
 |
| | Fig. 9 Fluorescence intensity (at 445 nm) changes of L–2Zn2+ (10 μM) in water solution (HEPES 10 mM, pH = 7.4) against various anions. The red bars represent the fluorescence intensity of L–2Zn2+ solution in the presence of 3.0 equiv. of miscellaneous anions; the gray bars represent the fluorescence intensity of the above solution upon further addition of 3.0 equiv. of S2−. λex = 315 nm. | |
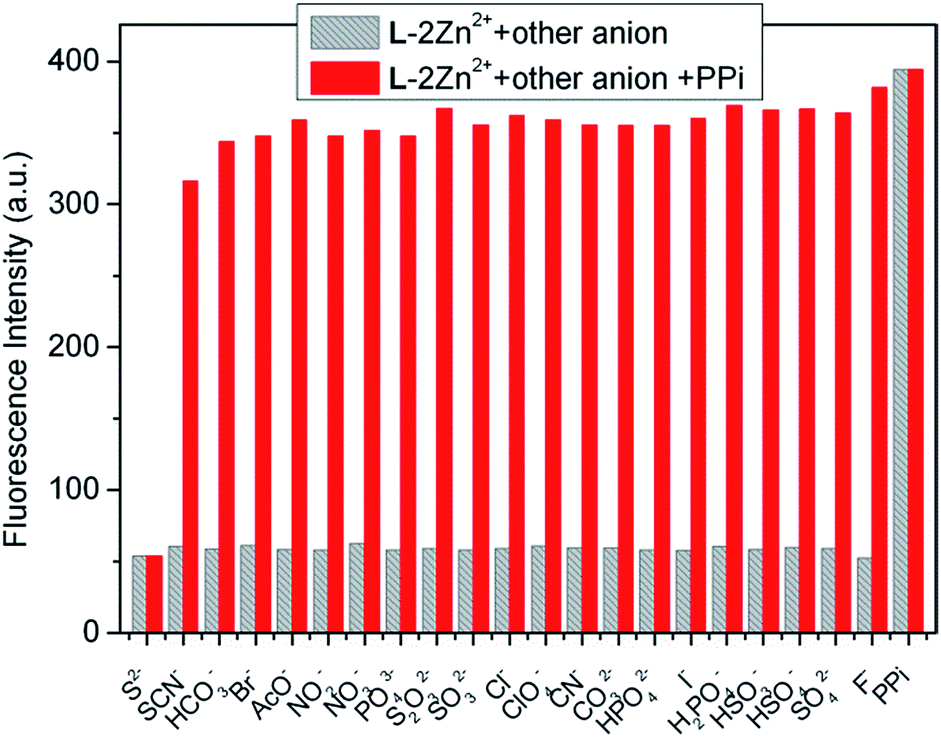
2.2.2 Recognition of PPi by L–2Zn2+ complex. As depicted in Fig. 7, addition of PPi to L–2Zn2+ solution results in the appearance of a blue-shifted emission band instead of recovery of the emission band of free L, which suggests that PPi may further bind with Zn2+ in L–2Zn2+ complex and hampered the ESIPT. To get further insights into the sensing behavior of the complex to PPi, fluorescence titration experiments were conducted (Fig. 10). On stepwise increase the added PPi concentration to L–2Zn2+ solution, the emission band centered at 445 nm gradually decreased and the newly formed emission band at 396 nm gradually increased. The fluorescence spectra changes terminated when 5 equiv. of PPi was employed.
 |
| | Fig. 10 Fluorescence spectra changes of the in situ generated L–2Zn2+ solution (10 μM, HEPES 10 mM, pH = 7.4) upon addition of increasing amounts of PPi (0 to 5.0 equiv.). λex = 315 nm. Inset: fluorescence color changes of L–2Zn2+ solution in the absence and presence of PPi under illumination with a portable UV lamp at 254 nm. | |
Non-linear least squares fitting of the titration profile based on a 1:1 binding equation provided a nice non-linear curve (Fig. S8†), which supports the 1:1 binding stoichiometry of PPi and L–2Zn2+. The association constant of PPi with L–2Zn2+ was estimated to be 1.11 × 105 M−1. Another powerful evidence for this binding ratio was obtained from the HRMS analysis of L–2Zn2+ solution in the presence of PPi. The prominent peak appeared at m/z 1057.0739 is assignable to [L + 2Zn2+ + P2O74− + Na+]+ (Fig. S9†), which supports not only the 1:2 binding ratio of L with Zn2+, but also the 1:1 binding stoichiometry of L–2Zn2+ and PPi. The plausible binding mode of L–2Zn2+ and PPi is illustrated in Scheme 2. Based on the titration data, the detection limit of L–2Zn2+ to PPi was evaluated to be 7.27 × 10−6 M utilizing the aforementioned method (Fig. S10†). Similarly, fluorescence competitive experiments were also examined to assay the anti-interference ability of PPi recognition (Fig. 11). The results demonstrate that the PPi recognition process is hardly hampered by other coexisting anion except S2−.
 |
| | Scheme 2 Proposed binding mode of L–2Zn2+ with PPi. | |
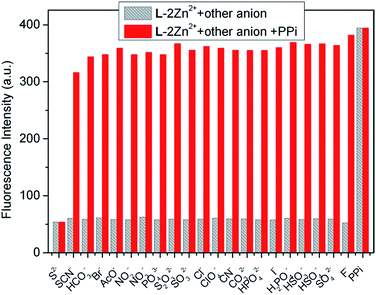 |
| | Fig. 11 Fluorescence intensity (at 396 nm) changes of L–2Zn2+ (10 μM) in water solution (HEPES 10 mM, pH = 7.4) to various anions (λex = 315 nm). The gray bars represent the fluorescence intensity of L–2Zn2+ solution in the presence of 5.0 equiv. of miscellaneous anions; the red bars represent the fluorescence intensity of the above solution upon further addition of 5.0 equiv. of PPi. | |
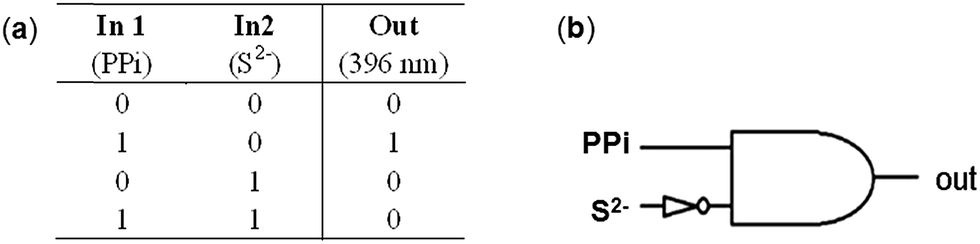
Recently, the development of molecular logic gates based on conversion of chemically encoded information into fluorescent signals has received considerable interest of chemists.3c,20 On the basis of the competition experiments described in Fig. 9 and 11, we noticed that the emission intensity of L–2Zn2+ solution restored to the emission state of free L in the presence of both PPi and S2− no matter the adding sequence of these two anions. Thus, an INHIBIT logic gate can be constructed when the fluorescence intensity of L–2Zn2+ solution at 396 nm was examined. Depending on the two chemical inputs, namely, input 1 (PPi) and input 2 (S2−), L–2Zn2+ can switch between high and low emission states. When the threshold value at 396 nm is set as 100, the output is recorded as 1 and 0 corresponding to the strong and weak emission intensity respectively. Input 1 elicits strong emission at 396 nm, represent as output ‘1’. On the contrary, input 2 results in a weak emission signal, represent as output ‘0’. Therefore, monitoring the emission intensity at 396 nm, upon addition of PPi and S2−, and their mixture results in an INHIBIT logic gate (Fig. 12).
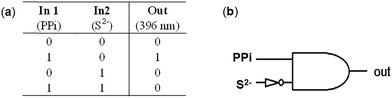 |
| | Fig. 12 Corresponding truth table of the logic gate (a) and illustration of the INHIBIT logic gate (b). | |
3. Conclusion
In summary, we have developed a new DPA dangled oxadiazole derivative L possessing ESIPT property for highly selective and ratiometric recognition of Zn2+. L binds two Zn2+ ions through its amide form, which can promote the ESIPT emission enhancement. The in situ generated L–2Zn2+ complex behaves relay recognition behavior to PPi and S2− via different response mechanisms. Addition of S2− to L–2Zn2+ solution results in Zn2+ sequestering and releasing of L, however, introducing PPi can lead to further complexation with L–2Zn2+. Thus, multi-analyte and relay recognition of L through modulating ESIPT has been achieved.
4. Experimental
4.1 Materials and instruments
Unless otherwise specified, solvents and reagents were of analytical grade from commercial suppliers and were used without further purification. Compounds 121 and 222 were prepared according to reported methods. 1H NMR and 13C NMR spectra were performed on an Agilent 400 MR spectrometer, and the chemical shifts (δ) were expressed in ppm and coupling constants (J) in Hertz. HRMS were measured on a Bruker micrOTOF-Q mass spectrometer (Bruker Daltonik, Bremen, Germany) or a matrix-assisted laser desorption ionization time-of-flight (MALDI-TOF) mass spectrometer (MALDI micro MX, Waters, USA). Fluorescence measurements were carried out on a Sanco 970-CRT spectrofluorophotometer (Shanghai, China). The pH measurements were made with a model PHS-25B meter (Shanghai, China).
4.2 Synthesis of sensor L
Dipicolyl amine (0.391 g, 1.96 mmol), potassium iodide (104.4 mg, 0.63 mmol) and 1.72 ml of diisopropylethylamine were added to 2 (0.4 g, 0.988 mmol) in 140 mL dry acetonitrile. The mixture was then heated to reflux and stirred for 10 h under nitrogen atmosphere. After removing the solvent, the residue was purified by column chromatograph with ethyl acetate and methanol (10:1, v/v) to afford 440 mg of sensor L. Yield: 60%. M.p. 101–102 °C. 1H NMR (400 MHz, DMSO-d6) δ 11.50 (s, 2H), 8.51 (d, J = 8.4 Hz, 2H), 8.38 (d, J = 4.4 Hz, 4H), 8.18 (d, J = 7.6 Hz, 2H), 7.63–7.54 (m, 10H), 7.33 (t, J = 7.6 Hz, 2H), 7.13 (t, J = 6.0 Hz, 4H), 3.87 (s, 8H), 3.44 (s, 4H); 13C NMR (100 MHz, DMSO-d6) δ 170.49, 162.86, 157.87, 149.13, 137.63, 137.04, 133.25, 129.41, 124.31, 123.83, 122.80, 121.44, 112.01, 60.43, 58.37; HRMS (MALDI-TOF-MS, positive mode) calcd for C42H38N10NaO3 753.3026 [L + Na+]+, found 753.2984.
4.3 General procedure for spectroscopic analysis
Doubly distilled water was used for all experiments. Sensor L was dissolved in HEPES (4-(2-hydroxyethyl)-1-piperazineethanesulfonic acid, 10 mM, pH = 7.4) buffered water to afford the test solution (10 μM). Titration experiments were carried out in 10 mm quartz cuvettes at 25 °C. Metal ions (as chloride or nitrate salts, 10 mM) or anions (as sodium or potassium salts, 10 mM) in water (HEPES 10 mM, pH = 7.4) were added to the host solution and used for the titration experiment.
Acknowledgements
We are grateful to the National Natural Science Foundation of China (no. 21476029, 21176029), the Natural Science Foundation of Liaoning Province (no. 20102004) and the Program for Liaoning Excellent Talents in University (no. LJQ2012096) for financial support.
Notes and references
-
(a) J. M. Berg and Y. Shi, Science, 1996, 271, 1081–1085 CAS;
(b) S. Y. Assaf and S. H. Chung, Nature, 1984, 308, 734–736 CrossRef CAS.
- C. F. Walker and R. E. Black, Annu. Rev. Nutr., 2004, 24, 255–275 CrossRef CAS PubMed.
-
(a) Z. Xu, J. Yoon and D. R. Spring, Chem. Soc. Rev., 2010, 39, 1996–2006 RSC;
(b) Z. Xu, K.-H. Baek, H. N. Kim, J. Cui, X. Qian, D. R. Spring, I. Shin and J. Yoon, J. Am. Chem. Soc., 2010, 132, 601–610 CrossRef CAS PubMed;
(c) Y.-P. Li, H.-R. Yang, Q. Zhao, W.-C. Song, J. Han and X.-H. Bu, Inorg. Chem., 2012, 51, 9642–9648 CrossRef CAS PubMed;
(d) Z. Xu, X. Liu, J. Pan and D. R. Spring, Chem. Commun., 2012, 48, 4764–4766 RSC;
(e) L. Xue, Q. Liu and H. Jiang, Org. Lett., 2009, 11, 3454–3457 CrossRef CAS PubMed.
- J. K. Heinonen, Biological Role of Inorganic Pyrophosphate, Kluwer Academic Publishers, Norwell, 2001 Search PubMed.
- S. Nussey and S. Whitehead, Endocrinology: An Integrated Approach; BIOS Scientific Publishers, Oxford, U.K., 2001 Search PubMed.
- M. Doherty, C. Belcher, M. Regan, A. Jones and J. Ledingham, Ann. Rheum. Dis., 1996, 55, 432–436 CrossRef CAS PubMed.
-
(a) W. Yu, J. Qiang, J. Yin, S. Kambam, F. Wang, Y. Wang and X. Chen, Org. Lett., 2014, 16, 2220–2223 CrossRef CAS PubMed;
(b) R. Villamil-Ramos, V. Barba Lopez and A. K. Yatsimirsky, Analyst, 2012, 137, 5229–5236 RSC;
(c) R. Kumar Pathak, V. Kumar Hinge, A. Rai, D. Panda and C. Pulla Rao, Inorg. Chem., 2012, 51, 4994–5005 CrossRef PubMed;
(d) F. Huang, C. Cheng and G. Feng, J. Org. Chem., 2012, 77, 11405–11408 CrossRef CAS PubMed;
(e) W.-H. Chen, Y. Xing and Y. Pang, Org. Lett., 2011, 13, 1362–1365 CrossRef CAS PubMed;
(f) S. K. Kim, D. H. Lee, J.-I. Hong and J. Yoon, Acc. Chem. Res., 2008, 42, 23–31 CrossRef PubMed.
- Hydrogen Sulfide, World Health Organization, Geneva, 1981, (Environmental Health Criteria, no. 19) Search PubMed.
- S. A. Patwardhan and S. M. Abhyankar, Colourage, 1988, 35, 15–18 CAS.
-
(a) Y. Yang, C. Yin, F. Huo, Y. Zhang and J. Chao, Sens. Actuators, B, 2014, 203, 596–601 CrossRef CAS PubMed;
(b) C. Liu, H. Wu, B. Han, B. Zhu and X. Zhang, Dyes Pigm., 2014, 110, 214–218 CrossRef CAS PubMed;
(c) F. Zheng, M. Wen, F. Zeng and S. Wu, Sens. Actuators, B, 2013, 188, 1012–1018 CrossRef CAS PubMed;
(d) C. Kar, M. D. Adhikari, A. Ramesh and G. Das, Inorg. Chem., 2013, 52, 743–752 CrossRef CAS PubMed;
(e) A. H. Gore, S. B. Vatre, P. V. Anbhule, S.-H. Han, S. R. Patil and G. B. Kolekar, Analyst, 2013, 138, 1329–1333 RSC;
(f) C. Yu, X. Li, F. Zeng, F. Zheng and S. Wu, Chem. Commun., 2013, 49, 403–405 RSC;
(g) M.-Q. Wang, K. Li, J.-T. Hou, M.-Y. Wu, Z. Huang and X.-Q. Yu, J. Org. Chem., 2012, 77, 8350–8354 CrossRef CAS PubMed;
(h) H. Peng, Y. Cheng, C. Dai, A. L. King, B. L. Predmore, D. J. Lefer and B. Wang, Angew. Chem., Int. Ed., 2011, 50, 9672–9675 CrossRef CAS PubMed.
- L. Tang, M. Cai, P. Zhou, J. Zhao, K. Zhong, S. Hou and Y. Bian, RSC Adv., 2013, 3, 16802–16809 RSC.
-
(a) M. Kumar, R. Kumar and V. Bhalla, Org. Lett., 2011, 13, 366–369 CrossRef CAS PubMed;
(b) P. Saluja, N. Kaur, N. Singh and D. O. Jang, Tetrahedron, 2012, 68, 8551–8556 CrossRef CAS PubMed;
(c) W. Wu, Z. Sun, Y. Zhang, J. Xu, H. Yu, X. Liu, Q. Wang, W. Liu and Y. Tang, Chem. Commun., 2012, 48, 11017–11019 RSC;
(d) M. Kumar, N. Kumar and V. Bhalla, Chem. Commun., 2013, 49, 877–879 RSC.
-
(a) J. Wu, W. Liu, J. Ge, H. Zhang and P. Wang, Chem. Soc. Rev., 2011, 40, 3483–3495 RSC;
(b) J. Zhao, S. Ji, Y. Chen, H. Guo and P. Yang, Phys. Chem. Chem. Phys., 2012, 14, 8803–8817 RSC.
-
(a) L. Tang, M. Cai, P. Zhou, J. Zhao, Z. Huang, K. Zhong, S. Hou and Y. Bian, J. Lumin., 2014, 147, 179–183 CrossRef CAS PubMed;
(b) S. Goswami, A. Manna, S. Paul, A. K. Das, P. K. Nandi, A. K. Maity and P. Saha, Tetrahedron Lett., 2014, 55, 490–494 CrossRef CAS PubMed;
(c) S. Goswami, A. Manna, S. Paul, A. K. Das, K. Aich and P. K. Nandi, Chem. Commun., 2013, 49, 2912–2914 RSC.
- L. Tang, X. Dai, K. Zhong, D. Wu and X. Wen, Sens. Actuators, B, 2014, 203, 557–564 CrossRef CAS PubMed.
-
(a) J. Wang, X. Liu and Y. Pang, J. Mater. Chem. B, 2014, 2, 6634–6638 RSC;
(b) A. K. Mahapatra, S. K. Manna, C. D. Mukhopadhyay and D. Mandal, Sens. Actuators, B, 2014, 200, 123–131 CrossRef CAS PubMed;
(c) S. Kim, M. S. Eom, S. K. Kim, S. H. Seo and M. S. Han, Chem. Commun., 2013, 49, 152–154 RSC;
(d) S. Yang, G. Feng and N. H. Williams, Org. Biomol. Chem., 2012, 10, 5606–5612 RSC;
(e) L. Tang, M. Cai, P. Zhou, J. Zhao, K. Zhong, S. Hou and Y. Bian, RSC Adv., 2013, 3, 16802–16809 RSC.
- N. Niamnont, N. Kimpitak, G. Tumcharern, P. Rashatasakhon and M. Sukwattanasinitt, RSC Adv., 2013, 3, 25215–25220 RSC.
- W. Lin, L. Yuan, Z. Cao, Y. Feng and L. Long, Chem.–Eur. J., 2009, 15, 5096–5103 CrossRef CAS PubMed.
- Y. Cai, X. Meng, S. Wang, M. Zhu, Z. Pan and Q. Guo, Tetrahedron Lett., 2013, 54, 1125–1128 CrossRef CAS PubMed.
-
(a) V. García, A. Fernández-Lodeiro, R. Lamelas, A. Macías, R. Bastida, E. Bértolo and C. Núñez, Dyes Pigm., 2014, 110, 143–151 CrossRef PubMed;
(b) M. Zhang, H.-N. Le, X.-Q. Jiang and B.-C. Ye, Chem. Commun., 2013, 49, 2133–2135 RSC.
- C. Incarvito, A. L. Rheingold, C. J. Qin, A. L. Gavrilova and B. Bosnich, Inorg. Chem., 2001, 40, 1386–1390 CrossRef CAS PubMed.
- A. Komaraiah, K. Ramakrishna, B. Sailu and P. S. N. Reddy, ARKIVOC, 2007, 14, 110–116 CrossRef.
Footnote |
| † Electronic supplementary information (ESI) available: 1H NMR, 13C NMR and HRMS of sensor L, and other supplementary data. See DOI: 10.1039/c4ra16347h |
|
| This journal is © The Royal Society of Chemistry 2015 |
Click here to see how this site uses Cookies. View our privacy policy here.