DOI:
10.1039/C4RA16346J
(Paper)
RSC Adv., 2015,
5, 15612-15620
Self-assembled serum albumin–poly(L-lactic acid) nanoparticles: a novel nanoparticle platform for drug delivery in cancer†
Received
14th December 2014
, Accepted 27th January 2015
First published on 28th January 2015
Abstract
We developed a new self-assembled bovine serum albumin–poly(L-lactic acid) nanoparticle platform for anticancer drug delivery made from a bovine serum albumin–poly(L-lactic acid) polymer conjugate. Depending on the ratio of bovine serum albumin (BSA) to poly(L-lactic acid) (PLLA), these conjugates self-assemble into uniform spherical nanoparticles with different sizes. Then, BA-loaded BSA–PLLA nanoparticles (BSA–PLLA/BA NPs) were prepared by using BSA–PLLA conjugates as prototype materials, and betulinic acid (BA) as a model drug. In vitro cytotoxicity studies with human lung cancer cell lines (A549) and murine Lewis lung carcinoma (LLC) cell lines suggested that the BSA–PLLA/BA NPs were significantly superior to the model drug BA in antitumor activity and the BSA–PLLA NPs were non-toxic. Compared to free BA, the BSA–PLLA/BA NPs provided significantly higher blood circulation half-time of free BA (5.02-fold). The antitumor effect of the BSA–PLLA/BA NPs in a mouse tumor xenograft model showed much better tumor inhibition efficacy and fewer side effects than that of free BA. It may be attributed to the preferential tumor accumulation and increases the solubility of the drug in water, strongly supporting their use as high-performance carriers for anti-cancer therapy.
1. Introduction
Polymeric nanoparticles composed of hydrophobic and hydrophilic segments are considered to be very promising nanocarriers for drug delivery owing to their small size, prolonged circulation time, and sustained drug release profiles.1 Generally, polymeric core–shell nanoparticles are assembled in an aqueous solution by using the hydrophobic interactions between core-forming segments.2 The hydrophilic outer shell provides colloidal stability, while the hydrophobic inner core serves as a container for hydrophobic drugs. Compared with other drug delivery systems, nanoparticles show their advantages in passive tumor targeting by vascular leakage via the enhanced permeability and retention (EPR) effect due to their size ranging from 10 to 200 nm, which is sufficiently small to avoid filtration by the spleen and lung, and big enough to allow extravasation into tumor tissue but not into normal tissue.3,4 Therefore, the use of polymeric nanoparticles for drug delivery is an effective strategy for passive tumor targeting.5,6
Modern drug delivery systems are evolving into crafted equipment with efficient cargo-loading capacities and multiple targeting moieties. Synthetic polymers have diverse chemical properties in terms of their size, composition, and have long been used to display multiple copies of functional units. In contrast to the synthetic counterparts, biopolymers such as viruses or virus-like particles exhibit uniform structure and size. Their surface, which molecular cloning and protein binding strategies, can accurately display various functional groups.7,8 Viruses infect specific cells within host organisms, replicate, damage the cells, and diffused from cell to cell in infectious cycles, thereby leading to disease.9 These viral characteristics have inspired designs and synthetic of various drug delivery systems10–12 which should mimic the structure and biological function of the virus surface proteins to make the drug easily absorbed by the target cells.
Albumin were considered to be efficiently absorbed by cells and specifically accumulated in tumor cells based on their interactions with some specific protein receptors in caveolae and caveolae-mediated endothelial transcytosis as well as on tumor cells.13,14 Thus, bovine serum albumin (BSA) could be utilized as a model protein as a ligand for tumor cell targeting. A type of drug-delivery vehicle that mimics both biological functions and surface structural properties of viruses is suggested to provide a new approach for improving the efficiency of the delivery of anti-cancer drugs into cancer cells. There are numerous examples of the modification of viral or non-viral protein structures with synthetic polymers using by chemical grafting approach.15–19 Although protein–hydrophilic polymer conjugate has been widely studied because of their interesting structures of amphiphilic molecular, however, as far as we know, the synthesis of protein–hydrophobic polymer conjugates with hydrophobic polymer in aqueous solution is still a challenge to the field.
In this study, we report a new drug delivery platform based on this type of unique nanoparticle which is self-assembled from a BSA–poly(L-lactide acid) (PLLA) conjugate. Albumin with biodegradable and biocompatible is a kind of nutrient to cells; PLLA, which is a non-toxic and compostable biopolymer to CO2 and water, is also approved by the US FDA medical applications. By a simple method for nanoparticles, core–shell spherical nanoparticles of BSA–PLLA were obtained with hydrophobic drugs being encapsulated. We have encapsulated betulinic acid (BA) as a model chemotherapy drug in the BSA–PLLA nanoparticles (NPs). BA was discovered in a National Cancer Institute drug screening program of natural plant extracts, and has been recognized to possess potent antitumor activity against ovarian carcinoma, breast, lung, and head/neck cancer.20–22 Until now, clinical application of BA in cancer therapy is limited due to the very lipophilic characteristics of BA and its consequently poor solubility, relatively short half-life, and low bioavailability.23–25 Here, the usefulness of BSA–PLLA NPs as a carrier of BA was investigated by measuring the loading efficiency, drug release, and cell cytotoxicity in vitro. We also examined the antitumor activity of the BSA–PLLA/BA NPs in tumor-bearing mice.
2. Experimental
2.1. Materials
L-Lactide (L-LA, 98%) was purchased from Tokyo Chemical Industry CO., LTD. Bovine serum albumin (BSA) and 4-dimethylaminopyridine (DMAP) were purchased from Sigma-Aldrich. Betulinic acid (BA) was purchased from Chengdu Preferred Biotechnology Co., Ltd. Vivaspin ultracentrifugation filters with 10 kDa MWCO were purchased from Fisher (Ottawa, ON, Canada). All other reagents were purchased from Sigma-Aldrich.
Penicillin and streptomycin, Gibco Dulbecco's Phosphate-Buffered Saline (DPBS), and Gibco Dulbecco's Modified Eagle's Medium (DMEM) were all bought from Invitrogen. Fetal bovine serum (FBS) was from HyClone. Cell-Counting Kit-8 (CCK-8) was supplied by the Dojindo Laboratories. Human lung cancer cells (A549) and murine Lewis lung carcinoma (LLC) cells were obtained from the Peking University Health Science Center (Beijing, China) and were cultured in the listed medium: A549 by RPMI 1640 with 10% FBS, 1% streptomycin–penicillin and LLC by DMEM with 10% FBS, 1% streptomycin–penicillin. All cell lines were maintained in an incubator supplied with a 5% CO2/95% air humidified atmosphere at 37 °C.
Female C57BL/6 mice, 6–7 weeks age, were purchased from Beijing HFK BIOSCIECE CO., Ltd. All animal experiments were performed in accordance with Guide for the Care and Use of Laboratory Animals, and approved by Experimental Animal Ethics Committee in Beijing.
2.2. Synthesis of BSA–PLLA conjugates
BSA (0.8 μmol) was dissolved in anhydrous dimethylsulfoxide (20 mL) stirring at 50 °C for 24 h. Then, the solution was centrifugated at 7000 rpm for 30 min to remove the insolubles. L-LA dissolved in the above solution at concentrations of 17.4 M, 4.2 M, or 0.3 M, followed by addition of EDC (0.1 M), NHS (0.1 mM), and DMAP (10 mM) with stirring to initiate the polymerization. The reaction was carried out for 24 h at 50 °C. Then the solution was precipitated with methanol–ethyl ether (1![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) :8, v/v) mixture. The precipitate was collected by centrifuge, washed with methanol–ethyl ether mixture for three times, dried in the vacuum desiccators (yield, 80%, 76%, and 68%, respectively). 1H-NMR was recorded on a Bruker DRX-600 Avance III spectrometer in DMSO-d6.
:8, v/v) mixture. The precipitate was collected by centrifuge, washed with methanol–ethyl ether mixture for three times, dried in the vacuum desiccators (yield, 80%, 76%, and 68%, respectively). 1H-NMR was recorded on a Bruker DRX-600 Avance III spectrometer in DMSO-d6.
2.3. Preparation of BSA–PLLA/BA nanoparticles (NPs)
Nanoparticles were prepared by a nanoprecipitation method as described previously.26 BSA–PLLA and BA in 0.2 mL of dry DMSO was mixed and added dropwise to a vortexing solution of 1.8 mL phosphate buffered saline (PBS) solution (pH 7.4) in a 5 mL conical tube. Vortexing was maintained for 1 min after solution addition. The resulting BSA–PLLA/BA NPs solutions were dialyzed against distilled water using a membrane with a molecular weight cutoff of 10 kDa for 3 h with two exchanges of dialysate, and the supernatant was filtered through a 0.8 mm membrane and lyophilized. The size of the particles was determined by dynamic light scattering with a particle analyzer (Zetasizer Nano-ZS, Malvern Instruments Ltd., Malvern, UK).
2.4. Determination of drug loading and in vitro drug release
BSA–PLLA/BA NPs (20 mg mL−1) was diluted into phosphate buffered saline adjusted to pH 7.4 and incubated at 37 °C. Aliquots were removed at different time points and filtered through 0.22 μm PVDF syringe filter. BA in the filtered was measured by high-performance liquid chromatography (HPLC, Waters) using a reverse phase column (C18). The detection was performed by using UV detector at 210 nm, 75:25 mixture (v/v) of acetonitrile–water as a mobile phase, flow rate at 1.0 mL min−1. The percentage was calculated on the basis of the ratio of the peak area of the sample at 0, 2, 6, 12, 24, 36 and 48 h vs. the initial area peak. Each stability profile represents the average of two independent runs with the same sampling schedules. The standard deviation of each point is typically 2% or less. Drug loading capacity (DLC) was calculated according to the following equation: DLC (%) = (weight of loaded BA/weight of BSA–PLLA/BA NPs) × 100%.
2.5. In vitro cell cytotoxicity tests
CCK-8 assay was used to evaluation the cell viability of different samples.27,28 Briefly, two types of lung cancer cells LLC and A549 cells were respectively seeded at a density of 3 × 103 and 4 × 103 cells per well, respectively, in 180 μL culture medium and incubated for 24 h. Then, the cells were treated with various samples (BA, BSA–PLLA, and BSA–PLLA/BA) at 37 °C in a humidified incubator with 5% CO2 for 72 h, where the samples of the BA were dissolved in dimethylsulfoxide (Merck, Darmstadt, Germany) and diluted into tissue culture medium before assay and BA dose ranged from 2.5 to 500 μg mL−1. 20 μL of CCK-8 solution was added to each well of the plate and incubated for another 1 h at 37 °C. Percent of cell viability can be calculated by measured the absence of the samples at 450 nm. The IC50 was calculated as polymer concentrations which inhibited growth of 50% of cells relative to non-treated cells according to Unger et al.29 IC50 was calculated using the Boltzmann sigmoidal function from Origin® 8.6 (OriginLab, Northampton, USA). Data are representative of three independent experiments.
2.6. In vitro hemolysis assay
The hemolytic activity of polymer solutions was investigated as reported earlier.30,31 Briefly, fresh blood samples were collected through cardiac puncture from rats. Ten milliliters of blood was added with EDTA-Na2 immediately to prevent coagulation. The red blood cells (RBCs) were collected by centrifugation at 1500 rpm for 10 min at 4 °C. After washing in ice-cold DPBS until the supernatant was clear, erythrocytes were diluted at a final concentration of 5 × 108 cells per mL in ice-cold DPBS. 1 mL BSA–PLLA, BSA–PLLA/BA or PEI25 K solution (1 mg mL−1 and 0.1 mg mL−1) was mixed with 1 mL erythrocyte suspension. DPBS and 1% Triton X-100 in DPBS were used as negative control (0% lysis) and positive control (100% lysis), respectively. Samples were incubated for 1 h at 37 °C under constant shaking. After centrifugation at 1500 rpm for 10 min at 4 °C, supernatant was analyzed for hemoglobin release at 541 nm using an infinite M200 microplate spectrophotometer (Tecan, Switzerland). Hemoglobin release was calculated as (ODsample − ODnegative control)/(ODpositive control − ODnegative control) × 100%. Hemolysis was determined from three independent experiments.
2.7. In vivo pharmacokinetic study in mice
12 tumor-free healthy C57BL/6 female mice were divided into two groups at random. Group 1 was treated with BA injection (20 mg kg−1), group 2 with BSA–PLLA/BA NPs (equal to 20 mg kg−1 BA and 148.2 mg kg−1 of whole BSA–PLLA/BA NPs, here the loading of BA in the whole BSA–PLLA/BA NPs is 13.55%), via the tail vein. After intravenous administration, blood samples were collected at 0.083, 0.25, 0.5, 1, 2, 5, 10, 24, 48, 72 h from the orbital plexus and centrifuged immediately at 3000 rpm for 10 min at 4 °C. The plasma was frozen at −20 °C until assay. To determine the level of total BA in each plasma sample, 100 μL of plasma was mixed with 50 μL of 0.1 N NaOH for 15 min in water bath at 37 °C, allowing the drug release. After that, 0.1 N HCl (50 μL) was added, followed by 100 μL methanol. After vortexed for 2 min, the mixture was sonicated for 5 min and centrifuged at 5000 rpm for 5 min. The clear supernatant was dried under nitrogen, reconstituted by 100 μL methanol before HPLC analysis. The HPLC employs a VYDAC 214TP54 (C18, 300A, 5 μm, 4.6 × 250 mm) with a UV detector, using a gradient of 15–100% of acetonitrile in 0.05% TFA at a flow rate of 1 mL min−1. Blood circulation data were plotted as the blood BA or BSA–PLLA/BA NPs levels with the unit of percentage of injected dose per gram (% ID per g) against time after injection.
2.8. In vivo antitumor efficacy test
Subcutaneous tumor xenograft models were established in the right axillary flank region of C57BL/6 female mice (6–7 weeks) by injecting 1 × 106 LLC cells in 200 μL DMEM medium per mouse. Treatments were initiated when tumors reached an average volume of 100 to 150 mm3, and this day was designated as day 0. On day 0, these mice were randomly divided into 3 groups (n = 6) and administered intravenous injection with PBS (control), free BA (10 mg kg−1), and BSA–PLLA/BA NPs (contains 10 mg kg−1 of BA and 74.1 mg kg−1 of whole BSA–PLLA/BA NPs, here the loading of BA in the whole BSA–PLLA/BA NPs is 13.55%), respectively, on days 0, 2, 4, 6, and 8. In the observation phase, mice were monitored for tumor sizes and body weights every day. Tumor volume was calculated using the formula: (L × W2)/2, where L is the longest and W is the shortest tumor diameter (millimeter). Relative tumor volume (RTV) was calculated at each measurement time point (where RTV was equal to the tumor volume at a given time point divided by the tumor volume prior to initial treatment). For efficacy studies, the percentage of tumor growth inhibition (%TGI) was calculated using the following formula: [(C − T)/C] × 100%, where C is the mean tumor volume of the control group at a specified time and T is the mean tumor volume of the treatment group at the same time. To monitor potential toxicity, we measured the weight of each mouse. For humane reasons, animals were killed and regarded as dead if the implanted tumor volume reached to 5000 mm3 or at the end of the experiment (>6 weeks). To further evaluate the hematological toxicity of BSA–PLLA/BA NPs, we collected 200 μL of blood of each mouse after final administration.
2.9. Detection of allergic reaction
Toxic side-effects of the current chemotherapeutical drugs are often causing a severe reduction in the quality of life, so the detection of allergic reaction is very necessary and important. Five groups of tumor-bearing mice (26–28 g, n = 6) were used in allergy testing studies (control, BA, BSA–PLLA NPs, and BSA–PLLA/BA NPs). The four samples were administrated via tail intravenous injection every two days at the BA dose of 10 mg kg−1 body weight. After administration with different samples for 10 days, orbit blood of mice in different groups was collected and centrifuged. Serum samples were analyzed according to the procedure of Mouse IgE ELISA.
2.10. Statistical analysis
All experiments in this study were performed at least three times, and the data were expressed as the mean standard deviation (SD). Statistical analyses were performed by analysis of variance (ANOVA). All statistical analyses were performed using a 95% confidence interval (p < 0.05).
3. Result and discussion
3.1. Synthesis of BSA–PLLA conjugates
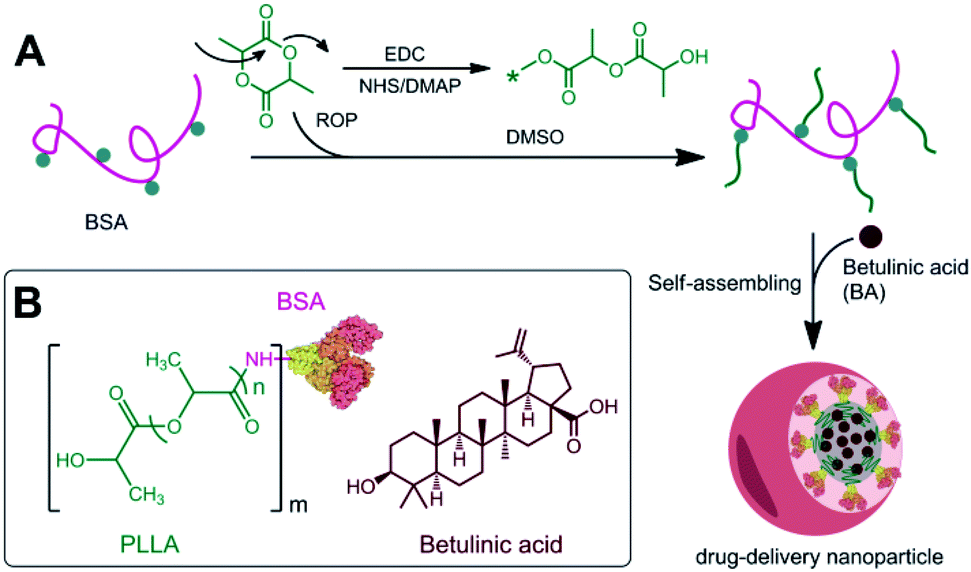
Protein–hydrophilic polymer conjugates have been widely studied and relative ease of synthesis in solution by chemical grafting approach.15–19,32 Due to the amphiphilic molecular structures, protein–hydrophobic polymer conjugates expected to have interesting additional functionalities. However, as far as we know, the protein–hydrophobic polymer conjugates has been difficult to synthetize mainly due to the insolubility of hydrophobic polymer in an aqueous solution. Here, the conjugate was successfully synthesized by BSA and hydrophobic PLLA (Scheme 1). BSA was dissolved in anhydrous dimethylsulfoxide (2 mg mL−1) at 50 °C. L-Lactide (L-LA) and 4-dimethylaminopyridine (DMAP) were added to conduct the ring opening polymerization (ROP) to form the BSA–PLLA conjugates at 50 °C. Additionally, in the ROP step of L-LA to PLLA using the EDC/NHS agents, carboxylic acid of homo-PLLA could be activated and conjugated on amino of BSA. 1H-NMR analysis (DMSO-d6) was determine the chemical structure of the conjugates (Fig. S1, ESI†). Depending on the feed ratio of L-LA to BSA, the mass fraction of PLLA in the conjugates were ∼25 wt%, ∼56 wt%, and ∼72 wt%.
 |
| | Scheme 1 (A) Synthesis of BSA–PLLA conjugate. (B) The chemical structure of BA and BSA–PLLA conjugate. | |
3.2. Nanoparticle formulation
The BSA–PLLA conjugate readily formed nanoparticles in aqueous solution. We have encapsulated betulinic acid (BA) as a model chemotherapy drug into the BSA–PLLA NPs (Scheme 1). BA is a kind of hydrophobic drug which can be loaded into the hydrophobic core of the nanoparticles.33–36 The conjugate in dimethylsulfoxide (2.5 mg mL−1) was dropwise into a PBS solution (pH 7.4) to prepare nanoparticles. The average size of the BSA–PLLA NPs increased from 94.25 to 103.36 to 162.21 nm with the increasing of the PLLA content from 25 to 56 to 72 wt% (Fig. 1A–C and Table 1), as determined by scanning electron microscopy (SEM), transmission electron microscopy (TEM), and dynamic light scattering (DLS). The weight ratio of BSA–PLLA of the conjugate in solution may be varied to control the size of nanoparticles (Table 1) which allowing us to selected the optimal properties of the BSA–PLLA NPs. The DLS measurements suggested that in aqueous solution aggregates are formed with high content of BSA while with 25 wt% of PLLA in the conjugate the nanoparticles are very well dispersed and readily more separate from each other. The BSA–PLLA NPs with 25 wt% of PLLA was chosen for the following drug loading.
 |
| | Fig. 1 SEM images and particle size distribution of BSA–PLLA NPs made with (A) 25 wt%, (B) 56 wt%, (C) 72 wt% PLLA, and (D) BSA–PLLA NPs with 25 wt% of PLLA loading with 13.55 ± 1.35 wt% BA. Scale bars are 100 nm. (E) The BA release from BSA–PLLA NPs (25 wt% PLLA) in PBS (pH 7.4) for different DLC of BA. | |
Table 1 Particle size and drug loading efficiency of nanoparticles
| Nanoparticle |
BSA–PLLA weight ratio |
BSA–PLLA/BA weight ratio |
Sizea (nm) |
DLCa (%) |
| Values are mean ± standard deviation (SD) (n = 3). |
| BSA–PLLA |
75/25 |
— |
94.25 ± 15.58 |
— |
| BSA–PLLA |
44/56 |
— |
103.36 ± 24.17 |
— |
| BSA–PLLA |
28/72 |
— |
162.21 ± 38.42 |
— |
| BSA–PLLA/BA |
75/25 |
5/1 |
118.32 ± 36.26 |
13.55 ± 1.35 |
| BSA–PLLA/BA |
75/25 |
3/1 |
156.25 ± 33.58 |
20.62 ± 1.28 |
DLC of BSA–PLLA/BA NPs was determined by HPLC and the results are shown in Fig. 1E. DLC was as high as 13.55% and 20.62% when BA was formulated in BSA–PLLA conjugates at a carrier/BA input ratio of 5/1 and 3/1 (w/w). Increasing the carrier/BA input ratios led to further increase in DLC and the nanoparticles sizes (Table 1). In addition, the drug-loaded nanoparticles exhibited bigger spherical shape than the blank ones (Fig. 1C and D). As shown in Fig. 1E, the BA-encapsulated nanoparticles with the loading ratio of 20.62 wt% occurs a initial burst of BA. In both formulations, BA was released from the BSA–PLLA NPs over 48 hours. When incubated with cancer cells, the BA release is possibly from the diffusion of BA molecules and the degradation of BSA–PLLA NPs, which are the two principal mechanisms of drug release from the nanoparticles.37 The BSA–PLLA/BA NPs with a 13.55 wt% loading ratio were selected as the lead candidate for the following tests in vitro and in vivo.
Furthermore, we observed the nanoparticles incubation in PBS solution (pH 7.4) to understand the self-assembly behavior. BSA–PLLA NPs and BSA–PLLA/BA NPs resulted in perfectly spherical particles both immediately after dispersing in PBS (Fig. 2A and C) and after incubating it in PBS for 2 days (Fig. 2B and D). In sharp contrast, a boundary of core–shell structure from the periphery of the nanoparticles was very clear presentation which was caused by the great difference in water solubility between BSA and PLLA parts. Most of the PLLA segments inner core serves as a container for BA, outer shell of the nanoparticles BSA chains improves the drug solubility and provides the colloidal stability.
 |
| | Fig. 2 TEM images of BSA–PLLA NPs dispersing in PBS immediately (A) and after incubating it in PBS for 2 days (B); BSA–PLLA/BA NPs dispersing in PBS immediately (C) and after incubating it in PBS for 10 days (D); scale bars are 200 nm. | |
3.3. In vitro cytotoxicity studies
To ensure the effective of the drug delivery before their entry into human application, in vitro cytotoxicity should be considered upfront.38,39 The cytotoxicity of BA, BSA–PLLA NPs, and BSA–PLLA/BA NPs to A549 or LLC cancer cells was evaluated using the CCK-8 assay. Cells were exposed to drug for 24, 48 or 72 h. Analysis of in vitro cytotoxicity measurements showed that the time-dependent cytotoxic effect of the BA solution was evident, which indicated 30.5% A549 and 41.6% LLC survival after 24 h, 14.3% A549 and 18.6% LLC survival after 48 h and 6.7% A549 and 9.0% LLC survival after 72 h at 500 μg mL−1 (equivalent to native BA) (Fig. 3A and B). To compare the potency of the BSA–PLLA/BA NPs, the concentrations of drug which killed 50% of the cells (IC50) were estimated from survival curves as shown in Fig. 3C and D, obtained from replicate experiments. The IC50 of BSA–PLLA/BA NPs was lesser than free drug. The BA carrying nanoparticles significantly increased the antitumor activities. The empty nanoparticles showed excellent biocompatibility. Around 110% cell survival was observed for cells treated with the empty nanoparticles, which might be due to that BSA–PLLA NPs contain BSA, the nutrient for cells.
 |
| | Fig. 3 Cellular cytotoxicity of BA, BSA–PLLA NPs, and BSA–PLLA/BA NPs. Cell viability of A549 (A) and LLC (B) cells treated with of BA, BSA–PLLA NPs, and BSA–PLLA/BA NPs (500 μg mL−1 equivalent to native BA, n = 3, error bars represent standard deviation). CCK-8 assay of BA, BSA–PLLA NPs, and BSA–PLLA/BA NPs with different concentration in A549 (C) and LLC (D) cell lines (n = 3, error bars represent standard deviation). | |
3.4. In vitro hemolysis studies
Detrimental interaction of conjugates with blood constituents such as red blood cells (RBCs) must be avoided when these conjugates are injected into the blood circulation as a carrier for drug delivery.30 Red blood cells were incubated with two concentrations of polymer as 1 mg mL−1 and 0.1 mg mL−1 for 1 h at 37 °C. Hemolysis was evaluated by measuring the amount of hemoglobin released in the supernatant at 541 nm (Fig. 4). Triton X-100 was used as positive control, which induced full hemoglobin release. BSA–PLLA/BA NPs at concentrations of 1 mg mL−1 and 0.1 mg mL−1 showed a comparable hemoglobin release to blank values (<5%), which was significantly lower than similar concentrations of PEI25 K, a cationic polymer known to have significant hemolytic effect.30 Despite BA was cytotoxic to the RBCs in a previous study,40 BSA–PLLA/BA NPs have been released little BA during the short incubation period (approximately 1 h), suggesting the excellent safety of BSA–PLLA/BA NPs.
 |
| | Fig. 4 In vitro hemolysis assay of BSA–PLLA NPs and BSA–PLLA/BA NPs compared to PEI25 K and Triton X-100 measured at 541 nm. Values are reported as the mean ± SD for triplicate samples. | |
3.5. In vivo pharmacokinetic studies
Long blood circulation half-time of a drug carrier is desired to improve the bioavailability of the drug. The determined drug concentration after release under basic condition was actually the total BA in plasma, the combination of both parent form and nanoparticle form. The plasma clearance curves of free BA and BSA–PLLA/BA NPs in mice were shown in Fig. 5. After intravenous administration of BA injection, the disappearance of BA from the blood circulation was very rapid with the plasma concentration below 20% of injected dose per gram (% ID per g) at 2 h. On the contrary, BSA–PLLA/BA NPs exhibited a remarkable prolonged clearance with the drug levels of 20% ID per g at 15 h after administration. The blood circulation half-time (t1/2) of free BA were 0.82 h. BSA–PLLA/BA NPs could extend the blood circulation half-time of BA from 0.82 h to 4.12 h, which were far longer (5.02-fold compared with BA) than values of BA.
 |
| | Fig. 5 Blood circulation curves and half-time of BSA–PLLA/BA NPs compared with free BA. Error bars were based on six mice per group at each time point. | |
3.6. In vivo anti-cancer efficacy
The results obtained above gave us a lot of motivated to explore the antitumor efficacy of BSA–PLLA/BA NPs in a mouse tumor model. In vivo antitumor efficiency of BSA–PLLA/BA NPs was tested by subcutaneous inoculation of LLC cells into mice. The size of BSA–PLLA/BA NPs was small enough in PBS solution (pH 7.4) that the nanoparticles were suitable for intravenous injection. Treatments with BA or BSA–PLLA/BA NPs given as multiple doses (q2d ×5) were initiated when tumors reached an average volume of 100 to 150 mm3. The results showed that, the effectiveness of the BSA–PLLA/BA NPs was significantly better than free BA. Multiple-dose treatment of BSA–PLLA/BA NPs caused 84.3% tumor growth inhibition (TGI) (on day 20), and by day 24, 83.3% of animals were survived. In contrast, multiple-dose free BA treatment resulted in 21.5% TGI (Fig. 6A, B and D and Table 2). Importantly, in line with the literature, no signs of systemic toxicity were observed by monitoring general behavior, appetite and mice body weight (Fig. 6C). When using the BA-encapsulated BSA–PLLA NPs for in vivo antitumor therapy, two effects are expected to increase the uptake of the BA-loaded nanoparticles by tumor cells: (1) the appropriate size (∼120 nm) can reduce renal clearance of the drug, and the drug is able to migrate through open malignant neovasculature and accumulate in tumor via the EPR effect;5,6,41 (2) BSA has many advantages such as low toxicity, excellent biocompatibility and biodegradability. BSA-based nanocarrier can offer the target agents and drugs with improved water solubility.42
 |
| | Fig. 6 Antitumor efficacy of BA and BSA–PLLA/BA NPs in LLC-bearing mice model. (A) Tumor volumes of mice with different treatment groups; dashed line: stop administration. (B) Therapeutic efficacy of different treatment. (C) The animal weights were recorded once per week and expressed over the 20 days observation. (D) Tumor photographs from each treatment group excised on day 20. | |
Table 2 LLC Xenograft Model (10 mg kg−1 BA, q2d ×5): efficacy comparison
| Compound |
Mean TV ± SDa (mm3) |
RTVa |
TGIa (%) |
Curesb (%) |
| Mean tumor volume (TV), RTV, and % TGI data were taken at day 20 (by day 20, a significant percentage of control animals were euthanized due to excess tumor burden). % cures were taken at day 24. |
| Control |
4375 ± 1950 |
35.0 ± 15.6 |
0 |
0 |
| BA |
2752 ± 1383 |
21.5 ± 10.8 |
37.1 |
16.7 |
| BSA–PLLA/BA NPs |
686 ± 383 |
5.2 ± 2.9 |
84.3 |
83.3 |
3.7. Evaluation of the side effects
Although BSA–PLLA/NPs NPs showed significant therapeutic effects in vivo, whether overcoming chemotherapy side effects is an important consideration for drug delivery by nanocarriers.43 To assess the therapeutic advantage of our BSA–PLLA NPs platform, we compared the blood chemistry of BA for the different treatment strategies. During the early development of the drugs, type I hypersensitivity is the most common type of the hypersensitivity reaction. Some of the natural anti-cancer drugs, such as paclitaxel, docetaxel, and teniposide cyclosporine, were typically associated with high morbidity of the type I hypersensitivity reaction. IgE antibodies have been demonstrated to play an important role in the mediation of type I hypersensitivity responses. So we choose IgE levels as the parameter for rapid evaluation of type I hypersensitivity reactions. The blood IgE levels of mice in different groups were shown in Fig. 7A. Compared to the control group, the mice treated with BA displayed a higher IgE level, which might be due to the poor water solubility. As expected, no significant change of IgE level was observed in the BSA–PLLA/BA NPs groups, which explored the idea that the use of these nanoparticles could dramatically reduce the risk of allergic reactions substantially. After treatment with different BA formulations, the blood of mice was also collected to test the white blood cell (WBC) count, which is typically used as an indicator of blood toxicity. The total WBC count of mice treated with free BA showed slightly lower than the normal group (Fig. 7B). No discernible decreases in WBC number of the mice treated with the nanoparticles were observed, indicating that the BSA–PLLA/BA NPs designed in this study could avoid severe hematotoxicity.
 |
| | Fig. 7 Subacute toxicities of different groups were reflected by IgE levels (A) and the WBC change (B) of mice. Data as means ± S.E.; n = 6. | |
4. Conclusions
In summary, a new protein–hydrophobic polymeric nanoparticle platform BSA–PLLA NPs has been prepared successfully. It is known that PLLA and BSA are both biocompatible and biodegradable in vivo. Through the antitumor studies in vitro and in vivo, we first demonstrated that the empty BSA–PLLA NPs had no toxicity to mice and good biocompatibility. In terms of loading capacity, a large amount of BA was well encapsulated into the BSA–PLLA NPs. Moreover, the BSA–PLLA/BA NPs shows excellent antitumor activity both in vitro and in vivo compared with free BA.
Acknowledgements
This study was funded by the Fundamental Research Funds for the Central Universities (200-1244951), the State Forestry Administration 948 Project of China (no. 2014-4-35), Beijing Natural Science Foundation of China (Grant no. 2142024), the National High Technology Research and Development Program of China (863 Program, no. 2014AA022109), and the National Natural Science Foundation of China (no. 20976179).
Notes and references
- L. Meng, W. Huang, D. Wang, X. Huang, X. Zhu and D. Yan, Biomacromolecules, 2013, 14, 2601–2610 CrossRef CAS PubMed.
- A. Harada and K. Kataoka, Prog. Polym. Sci., 2006, 31, 949–982 CrossRef CAS PubMed.
- Y. Matsumura and H. Maeda, Cancer Res., 1986, 46, 6387–6392 CAS.
- Z. Gao, A. N. Lukyanov, A. Singhal and V. P. Torchilin, Nano Lett., 2002, 2, 979–982 CrossRef CAS.
- H. Maeda, J. Controlled Release, 2012, 164, 138–144 CrossRef CAS PubMed.
- H. Maeda, J. Wu, T. Sawa, Y. Matsumura and K. Hori, J. Controlled Release, 2000, 65, 271–284 CrossRef CAS.
- N. F. Steinmetz, Nanomedicine: Nanotechnology, Biology and Medicine, 2010, 6, 634–641 CrossRef CAS PubMed.
- E. Strable and M. G. Finn, in Viruses and Nanotechnology, ed. M. Manchester and N. Steinmetz, Springer, Berlin Heidelberg, 2009, vol. 327, pp. 1–21 Search PubMed.
- P. De Vries, F. UytdeHaag and A. Osterhaus, J. Gen. Virol., 1988, 69, 2071–2083 CrossRef CAS.
- J. Zhu, J. Xue, Z. Guo, L. Zhang and R. E. Marchant, Bioconjugate Chem., 2007, 18, 1366–1369 CrossRef CAS PubMed.
- G. M. Kim, Y. H. Bae and W. H. Jo, Macromol. Biosci., 2005, 5, 1118–1124 CrossRef CAS PubMed.
- K. Babiuch, M. Gottschaldt, O. Werz and U. S. Schubert, RSC Adv., 2012, 2, 10427–10465 RSC.
- A. J. Montero, B. Adams, C. M. Diaz-Montero and S. Gluck, Expert Rev. Clin. Pharmacol., 2011, 4, 329–334 CrossRef CAS PubMed.
- F. Kratz, J. Controlled Release, 2008, 132, 171–183 CrossRef CAS PubMed.
- F. M. Brunel, J. D. Lewis, G. Destito, N. F. Steinmetz, M. Manchester, H. Stuhlmann and P. E. Dawson, Nano Lett., 2010, 10, 1093–1097 CrossRef CAS PubMed.
- A. J. Keefe and S. Jiang, Nat. Chem., 2012, 4, 59–63 CrossRef CAS PubMed.
- K. L. Heredia, D. Bontempo, T. Ly, J. T. Byers, S. Halstenberg and H. D. Maynard, J. Am. Chem. Soc., 2005, 127, 16955–16960 CrossRef CAS PubMed.
- J. Ge, D. Lu, J. Wang and Z. Liu, Biomacromolecules, 2009, 10, 1612–1618 CrossRef CAS PubMed.
- S. E. Averick, A. J. D. Magenau, A. Simakova, B. F. Woodman, A. Seong, R. A. Mehl and K. Matyjaszewski, Polym. Chem., 2011, 2, 1476–1478 RSC.
- X. Liu, I. Jutooru, P. Lei, K. Kim, S. O. Lee, L. K. Brents, P. L. Prather and S. Safe, Mol. Cancer Ther., 2012, 11, 1421–1431 CrossRef CAS PubMed.
- L. Li, Y. Du, X. Kong, Z. Li, Z. Jia, J. Cui, J. Gao, G. Wang and K. Xie, Clin. Cancer Res., 2013, 19, 4651–4661 CrossRef CAS PubMed.
- H. Ehrhardt, S. Fulda, M. Fuhrer, K. M. Debatin and I. Jeremias, Leukemia, 2004, 18, 1406–1412 CrossRef CAS PubMed.
- E. Pisha, H. Chai, I.-S. Lee, T. E. Chagwedera, N. R. Farnsworth, G. A. Cordell, C. W. W. Beecher, H. H. S. Fong, A. D. Kinghorn, D. M. Brown, M. C. Wani, M. E. Wall, T. J. Hieken, T. K. Das Gupta and J. M. Pezzuto, Nat. Med., 1995, 1, 1046–1051 CrossRef CAS.
- F. Li, R. Goila-Gaur, K. Salzwedel, N. R. Kilgore, M. Reddick, C. Matallana, A. Castillo, D. Zoumplis, D. E. Martin, J. M. Orenstein, G. P. Allaway, E. O. Freed and C. T. Wild, Proc. Natl. Acad. Sci. U. S. A., 2003, 100, 13555–13560 CrossRef CAS PubMed.
- S. M. Kamble, S. N. Goyal and C. R. Patil, RSC Adv., 2014, 4, 33370–33382 RSC.
- M. J. Ernsting, W.-L. Tang, N. MacCallum and S.-D. Li, Bioconjugate Chem., 2011, 22, 2474–2486 CrossRef CAS PubMed.
- H. Yue, W. Wei, Z. Yue, B. Wang, N. Luo, Y. Gao, D. Ma, G. Ma and Z. Su, Biomaterials, 2012, 33, 4013–4021 CrossRef CAS PubMed.
- W. Wei, P. P. Lv, X. M. Chen, Z. G. Yue, Q. Fu, S. Y. Liu, H. Yue and G. H. Ma, Biomaterials, 2013, 34, 3912–3923 CrossRef CAS PubMed.
- F. Unger, M. Wittmar and T. Kissel, Biomaterials, 2007, 28, 1610–1619 CrossRef CAS PubMed.
- R. Reul, J. Nguyen and T. Kissel, Biomaterials, 2009, 30, 5815–5824 CrossRef CAS PubMed.
- J. Nguyen, T. W. J. Steele, O. Merkel, R. Reul and T. Kissel, J. Controlled Release, 2008, 132, 243–251 CrossRef CAS PubMed.
- Y. Kang, C. Yang, P. Ouyang, G. Yin, Z. Huang, Y. Yao and X. Liao, Carbohydr. Polym., 2009, 77, 244–249 CrossRef CAS PubMed.
- B. Luo, S. Xu, A. Luo, W.-R. Wang, S.-L. Wang, J. Guo, Y. Lin, D.-Y. Zhao and C.-C. Wang, ACS Nano, 2011, 5, 1428–1435 CrossRef CAS PubMed.
- H. Yu, Z. Xu, D. Wang, X. Chen, Z. Zhang, Q. Yin and Y. Li, Polym. Chem., 2013, 4, 5052–5055 RSC.
- Y. Sanada, I. Akiba, K. Sakurai, K. Shiraishi, M. Yokoyama, E. Mylonas, N. Ohta, N. Yagi, Y. Shinohara and Y. Amemiya, J. Am. Chem. Soc., 2013, 135, 2574–2582 CrossRef CAS PubMed.
- X. Zhu, M. Fryd, B. D. Tran, M. A. Ilies and B. B. Wayland, Macromolecules, 2012, 45, 660–665 CrossRef CAS.
- S. Fredenberg, M. Wahlgren, M. Reslow and A. Axelsson, Int. J. Pharm., 2011, 415, 34–52 CrossRef CAS PubMed.
- C. Ge, J. Du, L. Zhao, L. Wang, Y. Liu, D. Li, Y. Yang, R. Zhou, Y. Zhao, Z. Chai and C. Chen, Proc. Natl. Acad. Sci. U. S. A., 2011, 108, 16968–16973 CrossRef CAS PubMed.
- Y. Zhang, S. F. Ali, E. Dervishi, Y. Xu, Z. Li, D. Casciano and A. S. Biris, ACS Nano, 2010, 4, 3181–3186 CrossRef CAS PubMed.
- M. Gao, P. M. Lau and S. K. Kong, Arch. Toxicol., 2014, 88, 755–768 CAS.
- J. Fang, H. Nakamura and H. Maeda, Adv. Drug Delivery Rev., 2011, 63, 136–151 CrossRef CAS PubMed.
- M. Roser, D. Fischer and T. Kissel, Eur. J. Pharm. Biopharm., 1998, 46, 255–263 CrossRef CAS.
- H. Zhang, Z. Ji, T. Xia, H. Meng, C. Low-Kam, R. Liu, S. Pokhrel, S. Lin, X. Wang, Y.-P. Liao, M. Wang, L. Li, R. Rallo, R. Damoiseaux, D. Telesca, L. Mädler, Y. Cohen, J. I. Zink and A. E. Nel, ACS Nano, 2012, 6, 4349–4368 CrossRef CAS PubMed.
Footnote |
| † Electronic supplementary information (ESI) available: 1H-NMR of BSA–PLLA with different PLLA content. See DOI: 10.1039/c4ra16346j |
|
| This journal is © The Royal Society of Chemistry 2015 |
Click here to see how this site uses Cookies. View our privacy policy here.