
 Open Access Article
Open Access Article This Open Access Article is licensed under a
This Open Access Article is licensed under a Creative Commons Attribution 3.0 Unported Licence
Electrochemistry for the generation of renewable chemicals: electrochemical conversion of levulinic acid†
Tatiane R.
dos Santos
ab,
Peter
Nilges
a,
Waldemar
Sauter
a,
Falk
Harnisch
b and
Uwe
Schröder
*a
aInstitute of Environmental and Sustainable Chemistry, Technische Universität Braunschweig, Hagenring 30, 38106 Braunschweig, Germany. E-mail: uwe.schroeder@tu-braunschweig.de; Fax: +49 5313918424; Tel: +49 5313918425
bDepartment of Environmental Microbiology, UFZ-Helmholtz Centre for Environmental Research, Permoserstrasse 15, 04318, Leipzig, Germany
First published on 9th March 2015
Abstract
The oxidative and reductive electrochemical conversion of levulinic acid to its primary products valeric acid, γ-valerolactone, 2,7-octanedione, 4-hydroxy-2-butanone and 3-buten-2-one is studied in detail. The reactions were performed in aqueous solutions and at ambient temperature, following the principles of green chemistry. The obtained primary reaction products were studied with respect to the oxidative and reductive electrochemical formation of secondary products, such as n-octane, 1-butanol and 1,3-butanediol. It is shown that the choice of electrolyte composition, educt concentration and the nature of the electrode material has a strong influence on the selectivity of product formation. For instance it is demonstrated that in alkaline solutions γ-valerolactone can be gained from levulinic acid at iron electrodes with similar Coulombic efficiency (∼20%) but higher selectivity (S = 70%) than on lead (S = 50%). Furthermore, for the first time the electrochemical two-step reaction of levulinic acid to 1-butanol via 4-hydroxy-2-butanone is reported. For some of the reaction pathways the main product is water insoluble, which allows a direct separation of the product and the potential electrolyte reuse in a semi-continuous process. Especially the use of the electrocatalytic hydrogenation may provide a path for the storage of electricity into liquid organic fuels as shown by a basic energetic assessment of all electrochemical conversions.
1. Introduction
Levulinic acid (LA), produced from lignocellulosic biomass, is considered to be an important precursor for renewable chemicals and liquid biofuels.1–3 The synthesis of levulinic acid from saccharose was reported already in 1840![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) 4 and since then has been studied from different perspectives. Generally, LA can be produced in high yields from carbohydrates and from lignocelluloses by acid catalyzed hydrolysis.5–15 The review by Rackemann and Doherty16 outlines the chemistry of levulinic acid synthesis and discusses current and potential technologies for producing levulinic acid from lignocellulosics biomass. Its molecular structure, comprising two functional groups (a keto and a carboxyl group), makes levulinic acid an ideal platform chemical. Thus, the lab-scale synthesis of bio-chemicals, polymers, pharmaceuticals, flavor agents, solvents, antifreeze agents, biofuels, plasticisers, and herbicides from LA has been reported.2,17–23 The respective syntheses are generally based on homogeneous and heterogeneous catalysis, whereas electrochemical conversions of LA are only rarely described. The electrochemical formation of γ-valerolactone (gVL) from LA was described by Tafel, when he studied the electrochemical reduction of levulinic acid in alkaline solution at a lead cathode.24–26 Further examples are the oxidative formation of 5-acetyl-2,9-decanedione in a methanol/water solution27 and recently the works of Xin et al. and Qiu et al. reporting on electrocatalytic processing of levulinic acid to produce biofuels.25,28
4 and since then has been studied from different perspectives. Generally, LA can be produced in high yields from carbohydrates and from lignocelluloses by acid catalyzed hydrolysis.5–15 The review by Rackemann and Doherty16 outlines the chemistry of levulinic acid synthesis and discusses current and potential technologies for producing levulinic acid from lignocellulosics biomass. Its molecular structure, comprising two functional groups (a keto and a carboxyl group), makes levulinic acid an ideal platform chemical. Thus, the lab-scale synthesis of bio-chemicals, polymers, pharmaceuticals, flavor agents, solvents, antifreeze agents, biofuels, plasticisers, and herbicides from LA has been reported.2,17–23 The respective syntheses are generally based on homogeneous and heterogeneous catalysis, whereas electrochemical conversions of LA are only rarely described. The electrochemical formation of γ-valerolactone (gVL) from LA was described by Tafel, when he studied the electrochemical reduction of levulinic acid in alkaline solution at a lead cathode.24–26 Further examples are the oxidative formation of 5-acetyl-2,9-decanedione in a methanol/water solution27 and recently the works of Xin et al. and Qiu et al. reporting on electrocatalytic processing of levulinic acid to produce biofuels.25,28
By means of an electrochemical hydrogenation coupled with a Kolbe reaction for the conversion of levulinic acid to n-octane, with valeric acid (VA) as intermediate product, we recently proposed the use of electrochemistry for the production of liquid biofuels (also denominated as “electrofuels”). The high selectivity (>95%) of the electrochemical hydrogenation of levulinic acid to valeric acid achieved in this study in batch-reactions was recently confirmed by Xin et al. for a flow cell process, yielding 90% valeric acid and 5% γ-valerolactone.25
In this work we investigate promising primary as well as secondary electrochemical conversion processes of levulinic acid, comprising oxidative as well as reductive pathways (Scheme 1). This study considerably expands the portfolio of chemicals that can be obtained from the electrochemical conversion of LA,29 as it introduces the electrochemical synthesis of not only valeric acid, and n-octane but also γ-valerolactone, 2,7-octanedione, 4-hydroxy-2-butanone and 3-buten-2-one (also known as methyl-vinyl-ketone) as well as n-octane, 1-butanol, 1,3-butanediol and methyl-tetrahydrofuran.
 | ||
| Scheme 1 Schematic illustration of the pathways for the primary and secondary electrochemical conversion of levulinic acid. (A) Electrochemical reduction of levulinic acid to: (I) γ-valerolactone or (II) valeric acid, (B) electrochemical oxidation from levulinic acid to: (III) 2,7-octanedione, (IV) 4-hydroxy-2-butanone or (V) 3-buten-2-one and (C) electrochemical conversion of the primary reaction products to: (VI) 2-methyltetrahydrofuran (electrochemical conversion was not reproducibly possible) (VII and VIII) n-octane, (IX) 1-butanol or (X) 1,3-butanediol. | ||
Scheme 1 summarizes the primary and secondary oxidation as well as reduction reactions investigated in this study. The derived products may either find direct application as fuels or solvents or may serve as intermediates for further processes – which are summarized in Table 1.
| Products | Applications | Production methods |
|---|---|---|
| Valeric acid (VA) | Intermediate to valeric fuels and additives | For example by catalytic hydrogenation of levulinic acid to valeric acid, typically performed in a two-step reaction at 150–350 °C, under hydrogen pressure of 10–40 bar and at platinum metal based catalysts3,31,32 |
| γ-Valerolactone (gVL) | Intermediate for butene, valeric acid, and 5-nonanone production. gVL can be used as renewable solvent | Catalytic hydrogenation using different transition metal based catalysis at high temperature (70–265 °C), under hydrogen atmosphere (5–150 bar)33–45 |
| 2,7-Octanedione (2,7-OD) | The occurring 2,7-octanedione can be used in a intramolecular aldol reaction46,47 or intramolecular pinacol coupling48 | Usually achieved by complex catalytic routes, e.g., based on Au catalyzed reactions of the di-acetylene at 120 °C,49 Ir-complex-catalyzed addition of water to 1,7-octadiyne at 70 °C (ref. 50) or the Pd catalyzed Wacker oxidation of olefins at 80 °C under oxygen (6 atm) in DMA51 |
| 4-Hydroxy-2-butanone (4H2B) | 4-Hydroxy-2-butanone is used as a platform chemical for further syntheses, like the dehydration to 3-buten-2-one52 and the reduction to 1,3 butanediol53 or to 1-butanol (electrochemical path shown below) | 4-Hydroxy-2-butanone is industrially produced by the coupling of fossil source based propanone and formaldehyde in basic media.54 The Wacker-type oxidation of 3-buten-1-ol,55 selective catalytic dehydrogenation of 1,3-butanediol,56–58 selective heterogeneous catalytic oxidation of 1,3-butanediol with H2O2 in CH3CN,59,60 and reduction of a dicarbony (1,3-butanone) at 130 °C over metallocene complexes61 are examples for the complex formation of 4-hydroxy-2-butanone62 |
| 3-Buten-2-one (MVK) | Used in the production of gas-tight plastics, a intermediate in the production of pharmaceutical, fungicides,63 vitamin A and vitamin E.64 Important compound in Michael-additions, Robinson-annulations, Diels–Alder-reactions, Baylis–Hillman-reactions, radical additions heterocycle-synthesis; often found in natural material synthesis (polyketides, macrolactone, terpenes, pheromones and alkaloids)63 | Technically, 3-buten-2-one is built from acetone with formaldehyde and diethylamine hydrochloride (Mannich-reaction) or by aldol-condensation of acetone and formaldehyde.63 The catalytic oxidation of levulinic acid over CuO/CeO2 and CuO/Al2O3 gives 2-buten-3-one in 15.5% yield at 175 °C (ref. 65) or 67.5% at 300 °C over CuO2,66 it is also patent-registered67 |
| 1,3-Butanediol (1,3-BDO) | Polyester plasticizers, humectants | |
| n-Octane | n-Octane is used as solvent, cleaning agent, fuel additive or as reaction agent like oxidation by cobalt68 aromatization69 is described also | Fractional distillation and refining of petroleum or the selective electrolytic reductive homocoupling of alkyl iodides at Ag–Pd cathodes in dipolar solvents70 |
| Methyl-tetrahydrofuran (MTHF) | 2-Methyltetrahydrofuran is mainly used as a higher boiling substitute for tetrahydrofuran as specialty solvent. 2-Methyltetrahydrofuran can also be used in the electrolyte formulation for secondary lithium electrodes and as a component of alternative fuels71,72 | 2-Methyltetrahydrofuran is produced by Ni-catalyzed hydrogenation of 2-methylfuran. This reaction can be performed in the liquid phase under H2 pressures of 2 MPa or in the vapor phase at atmospheric pressure with common supported Ni catalysts73 |
As Table 1 also shows, most of the conventional synthesis routes for the conversion of levulinic acid are high-temperature and high-pressure processes. Electrochemical synthesis may provide an alternative route, as the respective reactions can be carried out in aqueous solutions and at room temperature and thus fulfill major criteria of green chemistry.30
Additionally to the interesting product spectrum and its exploitation for (bio)chemical synthesis, the direct use of electric current for chemical conversions may even allow the storage of energy from renewable resources like wind power and photovoltaic in the form of chemical potential energy into compounds with higher energy density. For serving as a high energy density fuel molecule, oxygen functionalities have to be removed from LA, for which new processes are needed. For some of the conversions from LA to a (more reduced and thus higher energy dense) target molecule consecutive reactions, i.e. an oxidation followed by a reduction or vice versa, are necessary. Such a combined oxidation and reduction can, in principle, be achieved in one electrochemical cell.
As demonstrated in a principle energetic assessment of the described transformation routes, the electrochemical conversion of levulinic acid provides a promising platform.
With the results in the present manuscript, we aim to demonstrate that some well-known electrochemical reactions like the Kolbe and non-Kolbe reaction and the electrocatalytic hydrogenation, are well suitable for the conversion and generation of green platform chemicals.
2. Experimental section
2.1 Chemicals
All chemicals used in this study were of analytical grade. For qualitative and quantitative analysis, reference materials and solvents were used as purchased, without purification. 2,7-octanedione was produced by Kolbe-electrolysis and was purified by re-crystallization in heptane and its purity was confirmed by in-house NMR (1H and 13C) measurements.2.2 Electrode materials
As anode or cathode material were used: graphite foil (chemPUR, Germany), polycrystalline graphite sheet (SGL Carbon GmbH, Germany), graphite felt (SGL Carbon GmbH, Germany), lead sheet (99.999%, chemPUR, Germany), copper-sheet (99.999%, chemPUR, Germany), iron sheet (99.5%, chemPUR, Germany), nickel sheet (99.5% PHYWE Systeme GmbH & Co. KG, Germany), and platinum sheet (99.9%, chemPUR, Germany).A pretreatment procedure was applied to remove contaminants from the electrode surfaces. The lead electrodes were rinsed in 25% HNO3, treated with sand paper (grain size 8000) and washed with deionized water. Graphite felt electrodes were washed with deionized water and used directly. Platinum electrodes were pretreated by flame annealing and washed with deionized water. All the other electrodes were washed in HCl, treated with sand paper (grain size 8000) and washed with deionized water.
2.3 Electrochemical procedures
All electrochemical reactions were conducted under potentiostatic control using a potentiostat/galvanostat (AMEL 7050 (Amel srl, Milano, Italy) or a SP50 potentiostat (Bio-Logic SAS, Claix, France)). A three electrode configuration is used with EWE standing for the potential applied at the working electrode and ECE for potential measured at the counter electrode versus reference electrode, for which Ag/AgCl sat. KCl electrodes (SE11, Sensortechnik Meinsberg, Germany, 0.195 V vs. standard hydrogen electrode (SHE)) were used throughout the study. All electrode potentials in this study are reported versus Ag/AgCl sat. KCl, but normalized to standard hydrogen electrode for energetic calculations.As electrochemical cells for batch reaction one chamber undivided glass cells with 50 mL reaction volume and two-chamber H-type electrochemical glass cells with 40 mL anode and cathode chambers, separated via a cation exchange membrane (fumasep® FKE, Fumatech, Germany), were used, with a geometric electrode area varying from 5 to 24 cm2. All reported current densities are normalized to the geometric surface area of the working electrode.
The electrochemical flow cells were a custom-made Plexiglas® cell with 21 mL reaction volume and 11 cm2 anode and cathode area and a micro flow cell (Micro Flow Cell, ElectroCell, Denmark) with 10 mL volume and 10 cm2 for anode and cathode. The cells were operated in single-pass or recirculation mode at flow rates between 0.36 and 10 mL min−1 (thus the hydraulic residence time varies from 6 to 3600 s).
The duration of the electrolysis experiments varied according with initial concentration and achieved current. A detailed overview on all performed reactions is shown in the Tables SI 1 to SI 3 in the ESI.†
2.4 Analysis
Qualitative and quantitative analysis was carried out using gas chromatography-mass spectrometry (GC-MS) (Trace GC Ultra, DSQ II, Thermo Scientific, Germany) equipped with a TR-5MS (30 m × 0.25 mm ID × 0.25 μm film GC column Thermo Scientific, Germany) or TR-Wax MS (30 m × 0.25 mm ID × 0.25 mm film GC column, Thermo Scientific, Germany).Quantitative analysis was further carried out by gas chromatography (GC) with flame ionization detection (FID) (Hewlett Packard Series II 5890, Hewlett Packard, USA) equipped with a DB-5 (30 m × 0.25 mm ID × 0.25 mm film GC Column from Agilent JW Scientific, USA).
External calibration curves with different concentration levels in a range from 0 to 600 ng μL−1 were used for quantification.
Routine substance quantification was obtained by high performance liquid chromatography (HPLC), by means of a refractive index (RI) detector (Spectra system P4000, Finnigan Surveyor RI Plus Detector, Fisher Scientific) equipped with a HyperREZ XP Carbohydrate H + 8 μm (S/N: 026/H/012-227) column. Sulfuric acid (0.005 N, flow rate 0.5 mL min−1) served as the eluent. The column was operated at different temperatures depending on samples composition; the refractory index detector was operated at 40 °C.
Quantitative analysis was further realized with a HPLC-UV/VIS system (Hewlett Packard Series 1050, Hewlett Packard, United States of America) with a diode array detector (DAD) and equipped with a HyperREZ XP Carbohydrate H + 8 μm (S/N: 026/H/012-227). Phosphoric acid (0.05%, flow rate 0.5 mL min−1) served as the eluent. The column was kept at room temperature. Educt and product concentrations were determined using calibration curves in a range from 0 to 0.2 M.
For details on raw data (SI 1 for primary and SI 2 for secondary reactions) as well as calculations of Coulombic efficiency (CE) and selectivity (S) (SI 3) see ESI.†
3. Results and discussion
3.1 Primary reductive reactions: reduction of levulinic acid to valeric acid and γ-valerolactone
Scheme 2 shows the electrochemical reduction pathways of LA. Selected conditions have already been investigated by several authors, e.g., Xin et al.25 In the present study, we systematically studied the impact of a variety of electrode materials, the electrolyte composition and the educt concentration on the LA reduction. | ||
| Scheme 2 Reaction pathways of the electrochemical reduction of levulinic acid to (I) γ-valerolactone and (II) valeric acid (part A of Scheme 1). | ||
Fig. 1 summarizes the results of the levulinic acid reduction as a function of initial educt concentration. Changing the educt concentration does not affect the selectivity of the product formation: at a Pb electrode in 0.5 M H2SO4 electrolyte solution, and an applied working electrode potential of −1.8 V, valeric acid is always the main product. The increase of the initial educt concentration has, as expected, a positive impact to the Coulombic efficiency (CE) (see ESI S3† for definition). At sufficiently high levulinic acid concentration the hydrogenation process is the dominating reaction; whereas at low educt concentrations hydrogen evolution via water electrolysis as the main side reaction prevails, leading to low Coulombic efficiency and LA hydrogenation rate. Consequently, when using a LA starting concentration of 0.5 M instead of 0.05 M, the average conversion rate increases from 3.3 mg cm−2 h−1 to 29.5 mg cm−2 h−1 and the achieved Coulombic efficiency increases from 6% to 60%. A LA concentration of 0.5 M represents an optimum value, since a further increase of the starting concentration does not lead to a further significant increase of conversion rate and CE.
 | ||
| Fig. 1 Electrocatalytic hydrogenation of levulinic acid at Pb-electrode in a divided H-Cell at different initial concentration of levulinic acid after 4–8 hours electrolysis at a potential of −1.8 V. The formation of the organic product phase is shown in the inset picture. Details of all reactions can be found in Table SI 1† entry 1 to 17. | ||
Beside the Coulombic efficiency aspect, it is of special interest to achieve high product concentrations for the conversion of levulinic acid to valeric acid (Scheme 2 – route II), since spontaneous product separation can be achieved when the saturation concentration of valeric acid of 0.39 M is exceeded (see inset picture in Fig. 1).
Electrochemical conversions are heterogeneous processes, and as such, their mechanisms are decisively determined by the nature of the electrode material. Fig. 2 summarizes major parameters of the levulinic acid reduction as a function of the used electrode material and nature of electrolyte. In 0.5 M sulfuric acid, lead stands out clearly over the other electrodes in terms of educt conversion to valeric acid and Coulombic efficiency. This performance may be explained by the higher overpotential of the hydrogen evolution reaction at Pb-cathodes, since at Cu, Fe and Ni the water electrolysis commences already at an electrode potential of −1.0 V.
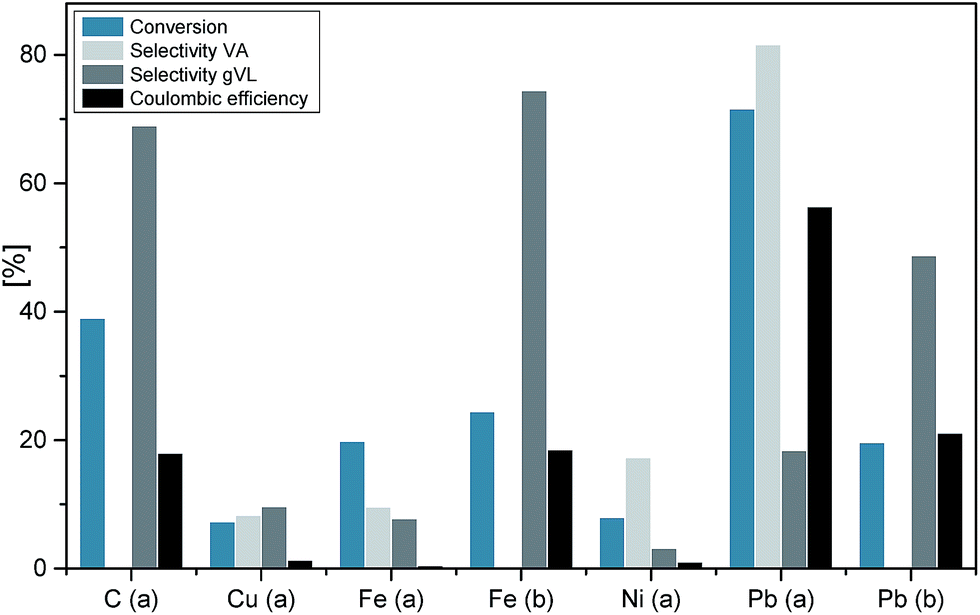 | ||
| Fig. 2 Electrocatalytic hydrogenation of levulinic acid in a divided H-Cell as a function of the nature of electrode material and electrolyte: (a) 0.5 M H2SO4 and (b) 1 M NaOH. The data are based on 2–4 hours electrolysis at an electrode potential of −1.8 V; the initial LA concentration was 0.5 M. | ||
When switching the cathode material from lead to carbon in acidic electrolyte, the hydrogenation of levulinic acid leads almost exclusively to the formation of γ-valerolactone (gVL). A conversion of 40% was obtained, which represents a ten-fold improvement as compared to the previous work by Xin et al.25 The selectivity of the gVL formation is 68%. Whereas gVL is formed in sulfuric acid solutions at Cu and Ni electrodes, no formation of valeric acid was observed in alkaline solution at this electrode materials. In alkaline solution gVL is the major product only when Pb and Fe electrodes are used. For the electrochemical conversion of LA to gVL at Pb electrodes a Coulombic efficiency of 21% can be achieved, whereas the use of Fe cathodes results in a slightly lower Coulombic efficiency (18%), but a higher selectivity. For this reason, and especially with regard to the sustainability of the process, Fe and C are very promising alternatives as electrode material for further investigations. It is very likely, that the Coulombic efficiency can be enhanced when using higher initial educt concentrations or when operating in a continuous system.
In summary, VA and gVL can both be obtained from LA with good selectivity and acceptable Coulombic efficiency.
3.2 Primary oxidative reactions: oxidation of levulinic acid to 2,7-octanedione, 4-hydroxy-2-butanone and 3-buten-2-one
Whereas the reductive conversion of levulinic acid may potentially be directly used for energy storage processes (see also 3.4.), the oxidation of levulinic acid is of primary interest for chemical production. Levulinic acid can be oxidized either to a keto-alkyl radical (one electron oxidation) or to a carbenium ion (two-electron oxidation) (see Scheme 3). Thereby the Kolbe product74–77 of LA, the dimer 2,7-octanedione, yields from the radical and its subsequent dimerization (route III).29 The carbenium ion, product of the two-electron oxidation, on the other hand, may react further either with nucleophiles like hydroxide ions (non-Kolbe-reaction or Hofer–Moest-reaction78–80), yielding 4-hydroxy-2-butanone (route IV), or may undergo an elimination of H+, forming 3-buten-2-one (route V).81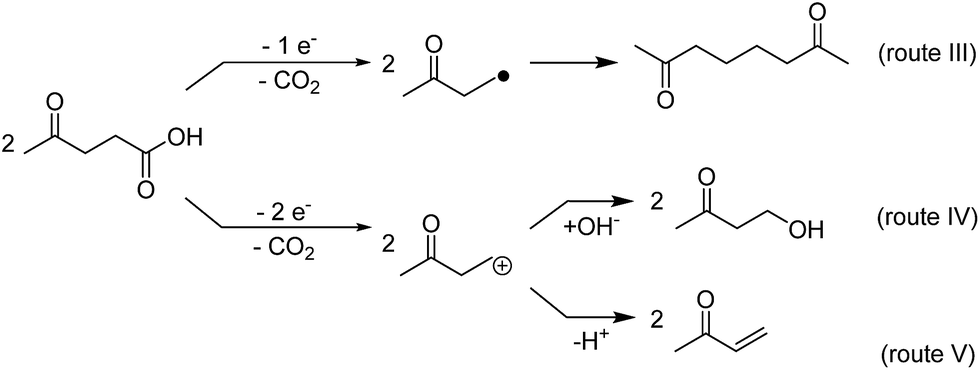 | ||
| Scheme 3 Reaction pathways of the electrochemical oxidation of levulinic acid. | ||
Interestingly, when performing the reaction in a membrane-separated flow cell, the major oxidation product is 3-buten-2-one (Scheme 3, route V). This finding (see Fig. S1†) can so far not be explained and has to be studied further.
3.3 Secondary electrochemical conversion steps
The products of the primary LA conversions can be used as educts for secondary electrochemical conversion steps (see Scheme 1 – secondary conversions). Some of these reactions, as the production of n-octane by the reduction of 2,7-octanedione and the Kolbe-coupling of valeric acid have been already reported.29 The following section presents further details of the Kolbe and the non-Kolbe reaction of valeric acid. Further, the hydrogenation of γ-valerolactone (Scheme 1, route VI) is very appealing, but unfortunately could not be experimentally proven. | ||
| Scheme 4 Reaction pathways for the electrochemical hydrogenation of 4-hydroxy-2-butanone. | ||
Similar as for the hydrogenation of levulinic acid, the Coulombic efficiency of the 4H2B hydrogenation can be significantly improved by higher initial educt concentration. Thus, the Coulombic efficiency of 1-butanol formation increased from 13% for a 0.08 M solution to 52% using a 0.5 M 4-hydroxy-2-butanone solution.
In order to achieve a higher selectivity of 1,3-butanediol formation, the hydrogenation of 4H2B was studied using different electrolytes and different electrode materials. Here, the use of phosphate buffer solution (0.5 M, pH 7) and iron electrodes yielded the highest 1,3-butanediol selectivity of 34% (see ESI, Table SI 2,† entry 1 to 7) (Scheme 4).
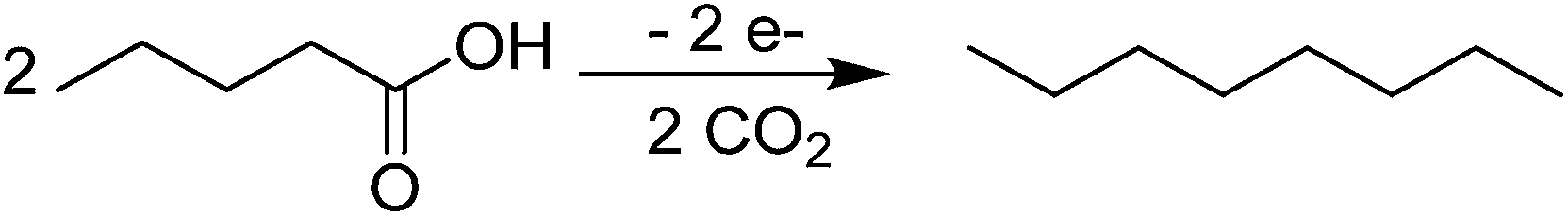 | ||
| Scheme 5 Reaction pathway for the electrochemical oxidation of VA to n-octane. | ||
As known from literature, the Kolbe reaction proceeds preferably at high current density and high educt concentration – both factors promoting the formation of high keto alkyl radical concentrations at the electrode surface and thus to the production of the dimer – in this case n-octane (Scheme 5). Additional to n-octane, this organic phase contains the side products butyl-valerate and 1-butanol.29 Wadhawan et al. proposed a possible mechanism for the production of esters during the Kolbe electrolysis in aqueous solution (Scheme 6).84Scheme 6 illustrates that butyl-valerate is formed, when only one CO2 is cleaved, whereas the formation of n-octane requires the complete elimination of both CO2.
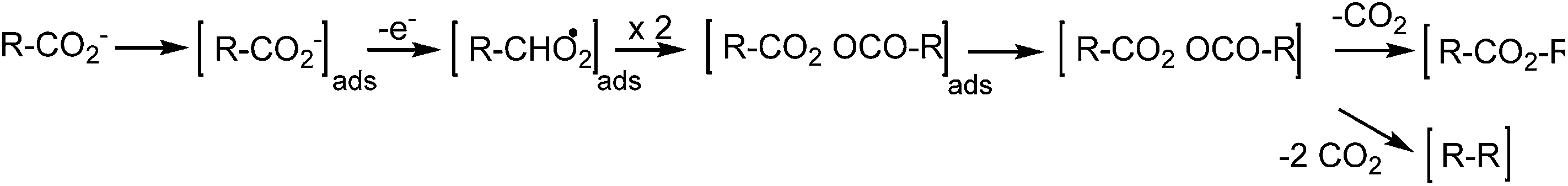 | ||
| Scheme 6 Potential mechanism for the formation of n-octane and butyl-valerate during Kolbe electrolysis (derived from Wadhawan84). | ||
The side product 1-butanol is generated via a Hofer–Moest reaction by a two electron oxidation of valeric acid and the formation of a carbenium ion that reacts with a hydroxide ion to form 1-butanol. The side product formation is dependent on the starting concentration of valeric acid and on the reactor configuration. In order to suppress this kind of side-reactions, experiments in a flow reactor were performed. Compared with our previous results for batch reactors, the Coulombic efficiency could not be improved with this setup, but even for a non-optimized system a CE of 50% was reached. A further improvement can be achieved, when using a flow reactor, as the selectivity of n-octane formation increases from 51 to 81%. The higher selectivity may result particularly from the lower residence time, which was for batch systems at least 4 hours, whereas a residence time not longer than 5 minutes, preventing further reactions, was realized in flow-through reactors.
 | ||
| Scheme 7 Anodic degradation of valeric acid at graphite electrodes. | ||
When the electrochemical conversion of short chain fatty acids, like valeric acid, is performed at carbon instead of platinum electrodes, the reaction leads to the formation of carbenium cations via a two-electron oxidation. These cations can then be attacked by a respective nucleophile (in water the hydroxide ion). Thus, the electrolysis of valeric acid at a carbon anode in aqueous environment (0.5 M H2SO4, see ESI, Table SI 3, entry 2) leads to the formation of alcohols. Depending on the location of the positive charge at the carbenium ion the hydroxide ion can attack either at position 1 (to form 1-butanol) or at position 2 (to form 2-butanol). At the applied positive anode potential a further oxidation of these products takes place: 1-butanol is oxidized to butanal, 2-butanol is oxidized to 2-butanone. Whereas ketones are inert to further oxidation, the aldehyde can be oxidized to carboxylic acids – which again can be decarboxylated in a further oxidation cycle. Thus – as illustrated in Scheme 7 – a successive shortening of the carbon chain occurs, leading to carbon dioxide as the final product.
In order to control the reaction and to prevent the successive decomposition, a charge transfer from a primary to secondary carbenium ion is necessary. The secondary carbenium ions will react with hydroxide ion to a secondary alcohol and thus no further oxidation than to the ketone is possible. Such control of the reaction did not succeed in full extent and the use of large electrolyte ions, such as ClO4− and HCO32−, as described in literature79 did not lead to the desired success. This cannot be fully explained, but the effect was mainly described for Pt-electrodes, where anion can adsorb thus prevent dimerization. In our study, we used graphite, at which anion adsorption may not, or only to a minor extent, take place.
As depicted in Fig. 3, the proposed in Scheme 7 degradation can be experimentally confirmed. Depending on the reaction time (and thus the degree of valeric acid conversion), the major reaction products and intermediates could be found.
 | ||
| Fig. 3 HPLC – chromatograms and the quantitative product analysis of a valeric acid (50 mM) non-Kolbe electrolysis (performed at a graphite electrode in 50 mM NaOH solution, at a potential of 2.5 V and a current density of 2.5 mA cm−2). | ||
The experiment also confirms the accumulation of 2-butanone – the product of the secondary carbenium ion. Interestingly, no acetone was detected throughout the experiments, which indicates that the primary propyl carbocation very quickly reacts to the primary alcohol, before the secondary carbenium ion is formed. The addition of other electrolyte species, however, did not have any significant impact on the course of the reaction (see Table SI 3†).
3.4 Energetic considerations for electrochemical levulinic acid conversions
Alternatively to their exploitation as platform chemicals (see Table 1) the electrochemical conversions of LA can be used for the storage of electric energy. The derived products may serve as alternative “green” fuels for combustion engines.85 To be exploited as an alternative fuel two key properties of a chemical have to be considered: (i) its ignition properties and (ii) its calorific value. Both properties are strongly influenced by the oxygen content of a substance. Dependent on the nature of the functional group, oxygen can be removed either reductively (via electrocatalytic hydration), or oxidatively (e.g., via Kolbe reaction). Certainly, a reduction reaction may represent a direct way to store electric energy into a chemical compound, whereas a (partial) oxidation lowers the absolute energy content with respect to the educt/starting material. Yet, the electrochemical oxidation reaction of levulinic acid (and its primary products) not only allows removing functional groups like carboxylic groups from a given educt (thus improving its ignition properties), but it also allows increasing the mass related energy density of the product by removing the oxygen containing functional groups. For instance the energy density of levulinic acid – a compound that is not suitable to be used in combustion engines due to the carboxylic group – of HLHV = 20.8 MJ kg−1 can be increased to that of 1-butanol (HLHV = 36 MJ kg−1) and of n-octane (HLHV = 44.4 MJ kg−1) by electrochemical oxidation (see Table SI 4†). (For the determination of the respective lower heating value, HLHV, the method according to Boie86 was used (see SI 3† for details)).In order to assess the energy efficiency of the reactions shown in Scheme 1, a calculation of the “energy storage efficiency”, i.e. the difference of the lower heating values of educt and product divided by the energy needed for the electrochemical reaction, for the different compounds and the input energy for the reaction is present in Table 2. Thereby the theoretical and experimentally required electric energy to drive the reaction(s) was compared to the increase in terms of energy density of the product molecules (see ESI S3† for details of definitions and all calculations.). Since in our study all reactions were performed in half-cell experiments, the energy calculation was based on the respective half-reaction, considering the needed average working electrode potential (vs. standard hydrogen electrode, SHE). The average working electrode potential for the reduction reactions was −1.8 or −1.5 V (vs. Ag/AgCl, corresponding respectively to a potential of −1.5 and −1.3 V vs. standard hydrogen electrode, SHE). For the oxidation reactions the applied working electrode potential varied from 2 to 10 V (vs. Ag/AgCl, corresponding respectively to a potential of 2.2 and 10.2 V vs. standard hydrogen electrode, SHE), please note that the higher oxidation potentials were only applied for systems using organic solvents.
| Route | Energy densitya (HLHV)/kJ mol−1 | Δ Molar heating values/kJ mol−1 | Achievedb | Theoretical maximum | ||||||||||
|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|
| zc | Average electrode potential/V | CE/% | Molar electric energy/kJ mol−1 | Energy storage efficiency/% | Average electrode potential/V | CE/% |
Molar electric energy/kJ mol−1 | Energy storage efficiency/% | ||||||
| a Estimated according to Boie.86 b Achieved in this study. c Number of electrons. | ||||||||||||||
| Educt | Product | |||||||||||||
| Reduction | I | Levulinic acid → γ-valerolactone | 2420 | 2650 | 230 | 2 | −1.5 | 20 | 1447 | 16 | −1.3 | 100 | 251 | 92 |
| II | Levulinic acid → valeric acid | 2420 | 2840 | 420 | 4 | −1.5 | 60 | 965 | 44 | −1.3 | 100 | 502 | 84 | |
| VI | γ-Valerolactone → 2-methyl-THF | 2751 | 2863 | 112 | 4 | — | — | — | — | −1.3 | 100 | 502 | 22 | |
| VIII | 2,7-Octanedione → n-octane | 4323 | 5047 | 724 | 8 | −1.5 | 10 | 10 526 | 7 | −1.3 | 100 | 1003 | 72 | |
| IX | 4-Hydroxy-2-Butanone → 1-butanol | 2083 | 2446 | 363 | 4 | −1.3 | 58 | 865 | 42 | −1.3 | 100 | 502 | 72 | |
| X | 4-Hydroxy-2-butanone → 1,3-butanediol | 2083 | 2273 | 190 | 2 | −1.3 | 2 | 12 543 | 2 | −1.3 | 100 | 251 | 76 | |
| Two steps | I + VI | Levulinic acid → 2-methyl-THF | 2420 | 2863 | 443 | — | — | — | — | 100 | 753 | 59 | ||
| II + VII | Levulinic acid → n-octane | 2420 | 5047 | 207 | 1795 | 11.53 | 100 | 743 | 28 | |||||
| III + VIII | Levulinic acid → n-octane | 2420 | 5047 | 391 | 11 071 | 3.53 | 100 | 1245 | 31 | |||||
| IV + IX | Levulinic acid → 1-butanol | 2420 | 2446 | 26 | 11 285 | 0.23 | 100 | 984 | 3 | |||||
| IV + X | Levulinic acid → 1,3-butanediol | 2420 | 2273 | −147 | 22 963 | −0.64 | 100 | 733 | −20 | |||||
Table 2 shows the maximum and so far achieved energy storage efficiencies. Regarding the one step reactions, only the reductions provide the possibility for energy storage (the values for the oxidations are shown in Table SI 5†). Most of the reduction reactions exhibit a potential for energy storage efficiency higher than 70%. Here, for the theoretically most favourable reduction of levulinic acid into γ-valerolactone, (with a maximum energy storage efficiency of 92%), only 16% are achieved in our experiments. However, clearly closer matches of the theoretical and achieved energy storage capacity are shown, e.g. for route II and route IX. For the two-step reactions the theoretical maximum values are lower and the achieved energy storage efficiencies are maximum ca. 10% (route II combined with route VII). These calculations show that electrochemical conversions, here of levulinic acid and its primary products, can provide an interesting and potentially energetically and thus economically competitive alternative not only for chemical production, but also for fuel generation and energy storage.
4. Conclusions
We have reported the electrochemical transformation of levulinic acid to its primary products valeric acid, γ-valerolactone, 2,7-octanedione, 4-hydroxy-2-butanol and 3-buten-2-one. In a secondary step, the obtained products were subjected to further electrochemical oxidations and reductions. The presented electrochemical approach represents an energetical and thus potentially economical alternative to the conventional catalytic processes. Thereby, the selectivity for the reaction products can be tailored by electrolyte composition, electrode material and current density. The use of aqueous electrolyte solutions, ambient pressure and room temperature allows obeying the rules of green chemistry. Therefore, the results demonstrate that renewable electricity could be used for the sustainable synthesis of a variety of renewable chemicals from levulinic acid and for the storage of electric energy in the form of organic compounds, partially being already suitable as drop-in biofuels.Acknowledgements
F.H. acknowledges support by the BMBF (Research Award “Next generation biotechnological processes – Biotechnology 2020+”) and the Helmholtz-Association (Young Investigators Group). This work was supported by the Helmholtz-Association within the Research Programme Renewable Energies. T.R.S acknowledges support by Albert und Anneliese Konanz-Stiftung (HS Mannheim).References
- T. W. a. G. Petersen, Top value added chemicals from biomass. Volume I: Results of Screening for Potential Candidates from Sugars and Synthesis Gas, Pacific Northwest National Laboratory (PNNL) and the National Renewable Energy Laboratory (NREL), 2004 Search PubMed.
- J. J. Bozell and G. R. Petersen, Green Chem., 2010, 12, 539–554 RSC.
- J. C. Serrano-Ruiz, D. Wang and J. A. Dumesic, Green Chem., 2010, 12, 574–577 RSC.
- G. J. Mulder, J. Prakt. Chem., 1840, 21, 203–240 CrossRef PubMed.
- P. Gallezot, Chem. Soc. Rev., 2012, 41, 1538–1558 RSC.
- A. Szabolcs, M. Molnar, G. Dibo and L. T. Mika, Green Chem., 2013, 15, 439–445 RSC.
- V. E. Tarabanko, M. Y. Chernyak, S. V. Aralova and B. N. Kuznetsov, React. Kinet. Catal. Lett., 2002, 75, 117–126 CrossRef CAS.
- J. Shen and C. E. Wyman, AIChE J., 2012, 58, 236–246 CrossRef CAS PubMed.
- B. Girisuta, L. P. B. M. Janssen and H. J. Heeres, Chem. Eng. Res. Des., 2006, 84, 339–349 CrossRef CAS.
- B. Girisuta, B. Danon, R. Manurung, L. P. B. M. Janssen and H. J. Heeres, Bioresour. Technol., 2008, 99, 8367–8375 CrossRef CAS PubMed.
- J. Hegner, K. C. Pereira, B. DeBoef and B. L. Lucht, Tetrahedron Lett., 2010, 51, 2356–2358 CrossRef CAS PubMed.
- B. Girisuta, L. P. B. M. Janssen and H. J. Heeres, Ind. Eng. Chem. Res., 2007, 46, 1696–1708 CrossRef CAS.
- H. Chen, B. Yu and S. Jin, Bioresour. Technol., 2011, 102, 3568–3570 CrossRef CAS PubMed.
- L. Peng, L. Lin, J. Zhang, J. Zhuang, B. Zhang and Y. Gong, Molecules, 2010, 15, 5258–5272 CrossRef CAS PubMed.
- R. Weingarten, W. C. Conner and G. W. Huber, Energy. Environ. Sci., 2012, 5, 7559–7574 CAS.
- D. W. Rackemann and W. O. S. Doherty, Biofuels, Bioprod. Biorefin., 2011, 5, 198–214 CrossRef CAS PubMed.
- D. J. Hayes, J. Ross, M. H. B. Hayes and S. Fitzpatrick, The biofine process–production of levulinic acid, furfural, and formic acid from lignocellulosic feedstocks, Wiley-VCH Verlag GmbH, Weinheim, Germany, 2008 Search PubMed.
- M. Kitano, F. Tanimoto and M. Okabayashi, Chem. Econ. Eng. Rev., 1975, 7, 25–29 CAS.
- R. Leonard, Ind. Eng. Chem., 1956, 48, 1330–1341 CrossRef.
- S. W. Fitzpatrick, in Feedstocks for the Future: Renewables for the Production of Chemicals and Materials, American Chemical Society, 2006, vol. 921, pp. 271–287 Search PubMed.
- D. C. Elliott and J. G. Frye, Patent: US 5,883,266, USA, 1999.
- G. W. Huber, S. Iborra and A. Corma, Chem. Rev., 2006, 106, 4044–4098 CrossRef CAS PubMed.
- V. Ghorpade and M. Hanna, in Cereals, ed. G. Campbell, C. Webb and S. McKee, Springer, USA, 1997, pp. 49–55 Search PubMed.
- J. Tafel and B. Emmert, Z. Elektrochem., 1911, 17, 569–572 Search PubMed.
- L. Xin, Z. Zhang, J. Qi, D. J. Chadderdon, Y. Qiu, K. M. Warsko and W. Li, ChemSusChem, 2013, 6, 674–686 CrossRef CAS PubMed.
- J. Tafel, Z. Elektrochem. Angew. Phys. Chem., 1911, 17, 569–572 Search PubMed.
- I. Cabasso, M. Li and Y. Yuan, RSC Adv., 2012, 2, 9998–10006 RSC.
- Y. Qiu, L. Xin, D. J. Chadderdon, J. Qi, C. Liang and W. Li, Green Chem., 2014, 16, 1305–1315 RSC.
- P. Nilges, T. R. dos Santos, F. Harnisch and U. Schröder, Energy. Environ. Sci., 2012, 5, 5231–5235 CAS.
- P. Anastas and N. Eghbali, Chem. Soc. Rev., 2010, 39, 301–312 RSC.
- J.-P. Lange, Patent: WO2009007391, 2009.
- J.-P. Lange, R. Price, P. M. Ayoub, J. Louis, L. Petrus, L. Clarke and H. Gosselink, Angew. Chem., Int. Ed., 2010, 49, 4479–4483 CrossRef CAS PubMed.
- H. Mehdi, V. Fábos, R. Tuba, A. Bodor, L. T. Mika and I. T. Horváth, Top. Catal., 2008, 48, 49–54 CrossRef CAS PubMed.
- Z.-p. Yan, L. Lin and S. Liu, Energy Fuels, 2009, 23, 3853–3858 CrossRef CAS.
- G. Braca, A. M. Raspolli Galletti and G. Sbrana, J. Organomet. Chem., 1991, 417, 41–49 CrossRef CAS.
- K. Osakada, T. Ikariya and S. Yoshikawa, J. Organomet. Chem., 1982, 231, 79–90 CrossRef CAS.
- Y. Gong, L. Lin and Z. Yan, Bioresearchs, 2011, 6, 686–699 CAS.
- L. E. Manzer, Appl. Catal., A, 2004, 272, 249–256 CrossRef CAS PubMed.
- W. Li, J.-H. Xie, H. Lin and Q.-L. Zhou, Green Chem., 2012, 14, 2388–2390 RSC.
- H. S. Broadbent, G. C. Campbell, W. J. Bartley and J. H. Johnson, J. Org. Chem., 1959, 24, 1847–1854 CrossRef CAS.
- R. V. Christian, H. D. Brown and R. M. Hixon, J. Am. Chem. Soc., 1947, 69, 1961–1963 CrossRef CAS.
- A. M. Hengne and C. V. Rode, Green Chem., 2012, 14, 1064–1072 RSC.
- P. Azadi, R. Carrasquillo-Flores, Y. J. Pagan-Torres, E. I. Gurbuz, R. Farnood and J. A. Dumesic, Green Chem., 2012, 14, 1573–1576 RSC.
- A. M. R. Galletti, C. Antonetti, V. De Luise and M. Martinelli, Green Chem., 2012, 14, 688–694 RSC.
- P. P. Upare, J.-M. Lee, D. W. Hwang, S. B. Halligudi, Y. K. Hwang and J.-S. Chang, J. Ind. Eng. Chem., 2011, 17, 287–292 CrossRef CAS PubMed.
- J.-P. Bouillon, C. Portella, J. Bouquant and S. Humbel, J. Org. Chem., 2000, 65, 5823–5830 CrossRef CAS PubMed.
- T. Hoffmann, G. Zhong, B. List, D. Shabat, J. Anderson, S. Gramatikova, R. A. Lerner and C. F. Barbas, J. Am. Chem. Soc., 1998, 120, 2768–2779 CrossRef CAS.
- D. S. Hays and G. C. Fu, J. Am. Chem. Soc., 1995, 117, 7283–7284 CrossRef CAS.
- P. Nun, R. S. Ramón, S. Gaillard and S. P. Nolan, J. Organomet. Chem., 2010, 696, 7–11 CrossRef PubMed.
- T. Hirabayashi, Y. Okimoto, A. Saito, M. Morita, S. Sakaguchi and Y. Ishii, Tetrahedron, 2006, 62, 2231–2234 CrossRef CAS PubMed.
- T. Mitsudome, T. Umetani, N. Nosaka, K. Mori, T. Mizugaki, K. Ebitani and K. Kaneda, Angew. Chem., Int. Ed., 2006, 45, 481–485 CrossRef CAS PubMed.
- N. Ichikawa, S. Sato, R. Takahashi and T. Sodesawa, Catal. Commun., 2005, 6, 19–22 CrossRef CAS PubMed.
- Y. Kobayashi, A. Matsuyama and T. Nikaido, Patent: US 5413922 A, 1995.
- J. T. Hays, G. F. Hager, H. M. Engelmann and H. M. Spurlin, J. Am. Chem. Soc., 1951, 73, 5369–5373 CrossRef CAS.
- S. Bawaked, Q. He, N. F. Dummer, A. F. Carley, D. W. Knight, D. Bethell, C. J. Kiely and G. J. Hutchings, Catal. Sci. Technol., 2011, 1, 747–759 Search PubMed.
- Y. Su, Y.-M. Liu, L.-C. Wang, M. Chen, Y. Cao, W.-L. Dai, H.-Y. He and K.-N. Fan, Appl. Catal., A, 2006, 315, 91–100 CrossRef CAS PubMed.
- S. Sato, R. Takahashi, H. Fukuda and K. Inui, J. Mol. Catal. A: Chem., 2007, 272, 164–168 CrossRef CAS PubMed.
- A. Kessat, A. Babadjamian and A. Iraqi, Eur. Polym. J., 2001, 37, 131–134 CrossRef CAS.
- X. Lang, Z. Li and C. Xia, Synth. Commun., 2008, 38, 1610–1616 CrossRef CAS PubMed.
- A. Bordoloi, S. Sahoo, F. Lefebvre and S. B. Halligudi, J. Catal., 2008, 259, 232–239 CrossRef CAS PubMed.
- T. Nakano, S. Umano, Y. Kino, Y. Ishii and M. Ogawa, J. Org. Chem., 1988, 53, 3752–3757 CrossRef CAS.
- J. Wang, Q. Wang, Y. Deng, Y. Li, B. H. Chen and R. Zhang, Appl. Catal., A, 2013, 452, 57–63 CrossRef CAS PubMed.
- U. Jahn, in Römpp Enzyklopädie Online, Georg Thieme Verlag KG, Stuttgart, 2014 Search PubMed.
- M. Eggersdorfer, G. Adam, M. John, W. Hähnlein, L. Labler, K.-U. Baldenius, L. von dem Bussche-Hünnefeld, E. Hilgemann, P. Hoppe, R. Stürmer, F. Weber, A. Rüttimann, G. Moine, H.-P. Hohmann, R. Kurth, J. Paust, H. Pauling, B. J. Weimann, B. Kaesler, B. Oster, U. Fechtel, K. Kaiser, B. de Potzolli, M. Casutt, T. Koppe, M. Schwarz, B.-J. Weimann, U. Hengartner, A. de Saizieu, C. Wehrli and R. Blum, in Ullmann's Encyclopedia of Industrial Chemistry, Wiley-VCH Verlag GmbH & Co. KGaA, 2000 Search PubMed.
- Y. Gong, L. Lin and B. Zhang, Chin. J. Chem., 2012, 30, 327–332 CrossRef CAS PubMed.
- Y. Gong, L. Lin, J. Shi and S. Liu, Molecules, 2010, 15, 7946–7960 CrossRef CAS PubMed.
- R. M. West and J. A. Dumesic, U.S. Pat. No. 7,960,592, 2010.
- F. Garcia-Ochoa, J. Querol and A. Romero, Ind. Eng. Chem. Res., 1990, 29, 1989–1994 CrossRef CAS.
- S. Trakarnroek, S. Jongpatiwut, T. Rirksomboon, S. Osuwan and D. E. Resasco, Appl. Catal., A, 2006, 313, 189–199 CrossRef CAS PubMed.
- P. Poizot, V. Jouikov and J. Simonet, Tetrahedron Lett., 2009, 50, 822–824 CrossRef CAS PubMed.
- V. R. Koch and J. H. Young, Science, 1979, 204, 499–501 CAS.
- M. G. Al-Shaal, A. Dzierbinski and R. Palkovits, Green Chem., 2014, 16, 1358–1364 RSC.
- H. E. Hoydonckx, W. M. Van Rhijn, W. Van Rhijn, D. E. De Vos and P. A. Jacobs, in Ullmann's Encyclopedia of Industrial Chemistry, Wiley-VCH Verlag GmbH & Co. KGaA, 2000 Search PubMed.
- H. J. Schäfer, Top. Curr. Chem., 1990, 152, 91–151 Search PubMed.
- A. K. Vijh and B. E. Conway, Fresenius. J. Anal. Chem., 1966, 224, 160–184 CrossRef.
- B. E. Conway and A. K. Vijh, Fresenius. J. Anal. Chem., 1966, 224, 149–159 CrossRef.
- H. J. Schäfer, Chem. Phys. Lipids, 1979, 44, 321–333 CrossRef.
- W. V. Miller and J. Hofer, Ber. Dtsch. Chem. Ges., 1894, 27, 461–470 CrossRef PubMed.
- H. Hofer and M. Moest, Justus Liebigs Ann. Chem., 1902, 323, 284–323 CrossRef CAS PubMed.
- E. Klocke, A. Matzeit, M. Gockeln and H. J. Schäfer, Chem. Ber., 1993, 126, 1623–1630 CrossRef CAS PubMed.
- D. J. Walton, ARKIVOC, 2002, 2002, 198–218 CrossRef.
- L. Eberson and K. Nyberg, Tetrahedron, 1976, 32, 2185–2206 CrossRef CAS.
- B. Wladislaw and H. Viertler, J. Chem. Soc. B, 1968, 576–579 RSC.
- J. D. Wadhawan, F. J. Del Campo, R. G. Compton, J. S. Foord, F. Marken, S. D. Bull, S. G. Davies, D. J. Walton and S. Ryley, J. Electroanal. Chem., 2001, 507, 135–143 CrossRef CAS.
- F. Harnisch, I. Blei, T. R. d. Santos, M. Moller, P. Nilges, P. Eilts and U. Schröder, RSC Adv., 2013, 3, 9594–9605 RSC.
- K. H. a. J. F. Grote, Dubbel-Taschenbuch für den Maschinenbau, Springer, Berlin Heideberg, New York, 2005 Search PubMed.
Footnote |
| † Electronic supplementary information (ESI) available. See DOI: 10.1039/c4ra16303f |
| This journal is © The Royal Society of Chemistry 2015 |