
Enhancing the electroluminescence performances of novel platinum(II) polymetallayne-based phosphorescent polymers through employing functionalized IrIII phosphorescent units and facilitating triplet energy transfer†
Zuan Huanga,
Boao Liua,
Jiang Zhaoa,
Yue Hea,
Xiaogang Yana,
Xianbin Xua,
Guijiang Zhou*a,
Xiaolong Yang*a and
Zhaoxin Wu*b
aMOE Key Laboratory for Nonequilibrium Synthesis and Modulation of Condensed Matter, State Key Laboratory for Mechanical Behavior of Materials, Department of Chemistry, Faculty of Science, Xi'an Jiaotong University, Xi'an 710049, P.R. China. E-mail: zhougj@mail.xjtu.edu.cn; Fax: +86-29-8266-3914
bKey Laboratory of Photonics Technology for Information School of Electronic and Information Engineering, Xi'an Jiaotong University, Xi'an 710049, P.R. China. E-mail: zhaoxinwu@mail.xjtu.edu.cn; Fax: +86-29-8266-4867
First published on 14th January 2015
Abstract
Novel orange phosphorescent polymers with platinum(II) polymetallayne-based backbones have been successfully developed through Sonogashira cross-coupling among bicarbazole moieties, functionalized IrIII phosphorescent blocks with electron injection/transporting (EI/ET) features, and trans-[PtCl2(PBu3)2]. Importantly, the very efficient energy-transfer process is observed from the triplet states of the polymetallayne-based backbone to the triplet metal-to-ligand charge transfer states (3MLCT) of the phosphorescent units in the polymer backbone, which will guarantee the high phosphorescent ability of these polymers. Benefiting from the weak conjugation-extending ability of the platinum(II) ions, the polymetallayne-based backbones show high triplet energy-level to effectively block the undesired reverse energy-transfer process. Furthermore, the EI/ET features of the functionalized IrIII phosphorescent units should balance the hole injection/transporting (HI/HT) of the bicarbazole moieties to improve the EL performances of these phosphorescent polymers. Benefiting from these merits, the phosphorescent polymers can furnish solution-processed phosphorescent OLEDs (PHOLEDs) with high EL efficiencies with current efficiency (ηL) of 9.17 cd A−1, external quantum efficiency (ηext) of 4.50% and power efficiency (ηP) of 4.04 lm W−1, representing very decent electroluminescent performances achieved by the orange phosphorescent polymers. This work herein might not only show the great potential of platinum(II) polymetallayne as the host segments in phosphorescent polymers, but also provide a new outlet to design and synthesise highly efficient phosphorescent copolymers.
1. Introduction
Phosphorescent polymers can show inherent advantages in fabricating high-performance phosphorescent organic light-emitting diodes (PHOLEDs) with a cheap solution-processed strategy. To prepare this kind of polymer, phosphorescent units, typically ppy-type iridium(III) complexes (ppy = 2-phenylpyridine anion), can be introduced to either the main chain1–5 or side chain6–11 of the conjugated or nonconjugated polymer backbones. Phosphorescent polymers with conjugated backbones can show advantages in promoting charge carrier injection/transporting in the emission layer (EML) of the PHOLEDs. The conjugated organic segments in the polymer backbone act as the host material for the phosphorescent units. In the operation of the PHOLEDs based on conjugated phosphorescent polymers, the majority of the formed excitons (ca. 75%) associated with the conjugated organic segments are triplet excitons, which transfer energy to the triplet states of the phosphorescent units to induce phosphorescent electroluminescence (EL). Clearly, the triplet energy-level of the conjugated backbones in the phosphorescent polymers should be higher than that of the phosphorescent units to guarantee the high efficiency of the EL process. Otherwise, there should be undesired reverse energy-transfer from the emissive triplet states of the phosphorescent units to the non-emissive triplet states of the conjugated backbones in the phosphorescent polymers, which would seriously impede the EL efficiencies. Hence, achieving high triplet energy-level of the polymer backbones is very crucial for developing high-performance phosphorescent polymers.Recently, platinum(II) polymetallaynes can show their great potential in various fields concerning opt-electronic applications, such as optical power limiting12,13 and photovoltaics,14,15 etc. Some bithiazole-based platinum(II) polymetallaynes can even show strong fluorescent to make them as suitable emitters for organic light-emitting diodes (OLEDs).16 All these important applications associated with polymetallaynes have benefited from their diverse photophysical behaviors which can be effectively manipulated through tuning the structure of organic spacers,17 i.e. aromatic alkynes. Due to the relatively weak conjugation extending ability associated with the platinum(II) ions, the concerned polymetallaynes generally show rather large band-gap (Eg) to afford triplet states with high energy-level.18–20 This character should render the great potential of the platinum(II) polymetallaynes as novel backbones for developing phosphorescent polymers. To the best of our knowledge, this potential associated with platinum(II) polymetallaynes has been hardly evaluated.
The high EL performances from the functionalized ppy-type iridium(III) phosphorescent emitters have clearly indicate their great potential in PHOLEDs. These phosphorescent emitters have been afforded with hole injection/transporting (HI/HT), electron injection/transporting (EI/ET), or even ambipolar features to benefit their EL performance.21–23 Bearing all these research achievements in mind, we have designed and prepared highly efficient conjugated phosphorescent polymers with both platinum(II) polymetallayne-type main chain and functionalized ppy-type iridium(III) complex with EI/ET features as orange phosphorescent units. Critically, the bicarbazole moieties with HI/HT characters have been employed as spacers of the polymetallayne main-chain to furnish triplet energy-level of the backbones and relieve the trouble of reverse energy-transfer. Together with the HI/HT properties of the bicarbazole moieties, the EI/ET features associated with the orange phosphorescent units can render the ambipolar features to the concerned polymetallayne-based phosphorescent polymers which can show high EL efficiencies.
2. Experimental section
2.1. General information
The commercially available chemicals were used directly as received. All reactions were preceded under inert atmosphere. The solvents were purified by standard methods under dry nitrogen before use. The reactions were monitored by thin-layer chromatography (TLC) with Merck pre-coated aluminum plates. Flash column chromatography and preparative TLC were carried out using silica gel for small molecular compounds. All Sonogashira copolymerization reactions were carried out with Schlenk techniques under nitrogen atmosphere.2.2. Physical characterization
1H NMR, 13C NMR and 31P NMR spectra were measured in CDCl3 with a BRUKER AVANCE III 400 MHz spectrometer. The chemical shifts were quoted relatively to the solvent residual peak at δ 7.26 for 1H and 77.0 for 13C, respectively. H3PO4 was used in the 31P NMR study as external reference. Fast atom bombardment (FAB) mass spectra were recorded on a Finnigan MAT SSQ710 system. UV-Vis spectra were recorded with a Shimadzu UV-2250 spectrophotometer. The photoluminescent (PL) properties of the copolymers were measured with an Edinburgh Instruments FLS920 fluorescence spectrophotometer. The lifetimes for the excited states were measured by a single photon counting spectrometer from Edinburgh Instruments FLS920 with a 360 nm picosecond LED lamp as the excitation source, while those at 77 K were obtained by with excitation from a xenon flash lamp. Differential scanning calorimetry (DSC) was performed with a NETZSCH DSC 200 PC unit under a nitrogen flow at a heating rate of 10 °C min−1. Thermogravimetric analysis (TGA) was conducted with a NETZSCH STA 409C instrument under nitrogen with a heating rate of 20 °C min−1. The molecular weights of the copolymers were determined by Waters 2695 GPC in THF. The weights were estimated by using a calibration curve of polystyrene standards. Cyclic voltammetry (CV) measurement for the sample solution was performed on Princeton Applied Research model 2273A potentiostat with a glassy carbon working electrode, a platinum counter electrode, and a platinum-wire reference electrode at a scan rate of 100 mV s−1. The solvent was deoxygenated dichloromethane, and the supporting electrolyte was 0.1 M [nBu4N][BF4]. Ferrocene (Fc) was added as an internal calibrant for the measurement, and all potentials reported were quoted with reference to the Fc/Fc+ couple. The polymer films on quartz substrate were obtained by spin-coating their chlorobenzene solution (ca. 20 mg mL−1) and their thickness was determined by Nanoview SE MF-1000 Ellipsometer. The PL spectra and lifetimes at 77 K were obtained by dipping the degassed sample CH2Cl2 solution in thin quartz tube into liquid nitrogen Dewar and recorded the data after standing 3 minutes.2.3. Synthetic details
The preparation details of the key compound L, monomer M-C, IrBr and the model polymer P-C were presented in the ESI.†![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) :1, v/v) as eluent to get the pure product as orange solid (0.29 g, 55%). 1H NMR (400 MHz, CDCl3, δ, ppm): 8.56 (s, 2H), 7.76 (s, 4H), 7.52 (d, J = 7.6 Hz, 2H), 6.79 (t, J = 7.2 Hz, 2H), 6.70 (t, J = 7.2 Hz, 2H), 6.25 (d, J = 7.6 Hz, 2H), 5.26 (s, 1H), 1.81 (s, 6H), 0.27 (s, 9H); 13C NMR (100 MHz, CDCl3, δ, ppm): 185.30, 165.65, 151.18, 148.82, 147.99, 141.51, 140.59, 140.22, 132.67, 130.91, 128.83, 127.46, 124.38, 120.26, 119.69, 119.14, 101.17, 100.87, 99.98, 28.55, −0.28; FAB-MS (m/z): 1072 [M]+. Elemental analysis calcd (%) for C49H47IrN2O6S2Si2: C 54.88, H 4.42, N 2.61; found: C 54.69, H 4.25, N 2.38.:1, v/v) as eluent to get the pure product as orange-red solid (0.035 g, 81%). 1H NMR (400 MHz, CDCl3, δ, ppm): 8.48 (s, 2H), 7.91 (dd, J = 8.4, 1.6 Hz, 2H), 7.84 (d, J = 8.8 Hz, 2H), 7.60 (d, J = 7.2 Hz, 4H), 7.55 (d, J = 8.4 Hz, 2H), 7.48 (t, J = 7.2 Hz, 2H), 7.38–7.31 (m, 8H), 5.28 (s, 1H), 3.38 (s, 2H), 1.80 (s, 6H); 13C NMR (100 MHz, CDCl3, δ, ppm): 185.35, 165.37, 151.3, 150.22, 147.62, 141.15, 140.46, 140.06, 132.77, 130.82, 128.88, 127.49, 124.56, 120.40, 119.30, 118.63, 101.64, 82.70, 79.00, 28.59; FAB-MS (m/z): 928 [M]+. Elemental analysis calcd (%) for C43H31IrN2O6O2: C 55.65, H 3.37, N 3.02; found: C 55.48, H 3.28, N 3.11.
:1, v/v) as eluent to get the pure product as orange solid (0.29 g, 55%). 1H NMR (400 MHz, CDCl3, δ, ppm): 8.56 (s, 2H), 7.76 (s, 4H), 7.52 (d, J = 7.6 Hz, 2H), 6.79 (t, J = 7.2 Hz, 2H), 6.70 (t, J = 7.2 Hz, 2H), 6.25 (d, J = 7.6 Hz, 2H), 5.26 (s, 1H), 1.81 (s, 6H), 0.27 (s, 9H); 13C NMR (100 MHz, CDCl3, δ, ppm): 185.30, 165.65, 151.18, 148.82, 147.99, 141.51, 140.59, 140.22, 132.67, 130.91, 128.83, 127.46, 124.38, 120.26, 119.69, 119.14, 101.17, 100.87, 99.98, 28.55, −0.28; FAB-MS (m/z): 1072 [M]+. Elemental analysis calcd (%) for C49H47IrN2O6S2Si2: C 54.88, H 4.42, N 2.61; found: C 54.69, H 4.25, N 2.38.:1, v/v) as eluent to get the pure product as orange-red solid (0.035 g, 81%). 1H NMR (400 MHz, CDCl3, δ, ppm): 8.48 (s, 2H), 7.91 (dd, J = 8.4, 1.6 Hz, 2H), 7.84 (d, J = 8.8 Hz, 2H), 7.60 (d, J = 7.2 Hz, 4H), 7.55 (d, J = 8.4 Hz, 2H), 7.48 (t, J = 7.2 Hz, 2H), 7.38–7.31 (m, 8H), 5.28 (s, 1H), 3.38 (s, 2H), 1.80 (s, 6H); 13C NMR (100 MHz, CDCl3, δ, ppm): 185.35, 165.37, 151.3, 150.22, 147.62, 141.15, 140.46, 140.06, 132.77, 130.82, 128.88, 127.49, 124.56, 120.40, 119.30, 118.63, 101.64, 82.70, 79.00, 28.59; FAB-MS (m/z): 928 [M]+. Elemental analysis calcd (%) for C43H31IrN2O6O2: C 55.65, H 3.37, N 3.02; found: C 55.48, H 3.28, N 3.11.2.4. OLED fabrication and measurements
The pre-cleaned ITO glass substrates were treated with ozone for 20 min. Then, the PEDOT:PSS was deposited on the surface of ITO glass by spin-coating method to form a 45 nm-thick hole-injection layer. After being cured at 120 °C for 30 min in the air, the emitting layer (35 nm) was obtained by spin-coating a chlorobenzene solution of each phosphorescent polymer. The ITO glass was dried in a vacuum oven at 50 °C for 20 min and it was transferred to the deposition system for organic and metal deposition. TPBi (45 nm), LiF (1 nm) and Al cathode (100 nm) were successively evaporated at a base pressure less than 10−6 Torr. The EL spectra and CIE coordinates were measured with a PR650 spectra colorimeter. The L–V–J curves of the devices were recorded by a Keithley 2400/2000 source meter and the luminance was measured using a PR650 SpectraScan spectrometer. All the experiments and measurements were carried out under ambient conditions.3. Results and discussion
3.1. Synthesis and characterization
The synthetic procedures for the phosphorescent polymers with platinum(II) polymetallayne-type main chain are shown in Scheme 1. The synthetic details for some key compounds required for preparation of the monomers M-C and M-Ir are provided in the ESI† (Scheme S1, ESI†). The precursor complex IrBr was firstly prepared according to the established two-step strategy by the cyclometalation of IrCl3·nH2O with the corresponding organic ligand L, followed by coordination of the acetylacetone (acac) anion in the presence of Na2CO3 (see ESI†). The compound IrSi was prepared by Sonogashira cross-coupling reaction between IrBr and trimethylsilylacetylene. Finally, the phosphorescent monomer M-Ir was obtained by cleaving of trimethylsilane groups from IrSi with [nBu4N]F. The MS and NMR spectra for these key compounds have been provided in ESI (Fig. S1†). It has been shown that the SO2Ph group can show electron injection/transporting (EI/ET) abilities.24–26 Hence, the functionalized phosphorescent monomer M-Ir together with the good hole injection/transporting (HI/HT) ability of the bicarbazole-based M-C might afford some ambipolar characters to the final phosphorescent polymers, benefiting their EL performances. | ||
| Scheme 1 Synthetic scheme for the orange phosphorescent polymers. | ||
The two alkynyl groups in both M-Ir and M-C can afford their ability of copolymerization with trans-[PtCl2(PBu3)2] to form the designed polymetallayne-type phosphorescent polymers, which have been prepared by Sonogashira cross-coupling procedure provided in Scheme 1. The feed ratio of the functionalized orange phosphorescent monomer M-Ir was set from 2.15 to 8.97 mol%, the corresponding weight percentage ca. 2.2, 4.3, 6.6, and 8.9 wt%. In the final stage of the polymerization, phenylacetylene was added as the end-capping regent. According to the feed ratios of M-C and M-Ir, the obtained copolymers are named P-SC-1 (m:n = 193:5), P-SC-2 (m:n = 183:11), P-SC-3 (m:n = 172:16), and P-SC-4 (m:n = 162:22), respectively (Scheme 1). In order to obtain samples in higher purity, the CuI catalyst was removed by filtration of the polymer solution with 0.45 μm PTFE syringe filter.
In the 1H NMR spectra for all the phosphorescent polymer, all the main resonance peaks with δ at ca. 8.39, 8.28, 8.21, 8.15 ppm together with the ones at ca. 7.82–7.76, 7.47–7.42 and 4.30 ppm are similar to those of the model polymer P-C (Fig. S2 in ESI†) due to the low content of the IrIII phosphorescent units. However, very weak signal at ca. 5.23 ppm can still be observed due to the acac ligand in the phosphorescent units. The broad of peak with ca. 2.26 ppm has been induced by the n-butyl groups in the chelated PBu3 moieties. All these spectral data have properly indicated that all the building blocks have been copolymerized together successfully. These phosphorescent polymers can possess very good processibility afforded by the n-butyl groups from both the bicarbazole blocks and platinum(II) units, since they are readily soluble in various common solvents, such as CH2Cl2, CHCl3, and THF etc. Taking polystyrene as standards, the number average molecular weights (Mn) of the copolymers range from 3.3 × 104 to 3.8 × 104 with polydispersity indices (PDIs) between 1.6 and 1.8.
In order to properly investigate the photophysical and electrochemical properties of the orange phosphorescent copolymers, the model platinum(II) polymetallayne P-C has also been obtained through Sonogashira cross-coupling between M-C and trans-[PtCl2(PBu3)2] (Scheme S1†). Owing to the low content of the IrIII phosphorescent units in these copolymers, the photophysical and electrochemical behaviors of the polymetallayne backbones in these phosphorescent polymers can be safely represented by the model polymer P-C.
3.2. Thermal properties and photophysical characters
The thermal properties of the platinum(II) polymetallayne-based phosphorescent polymers have been investigated by thermogravimetric analysis (TGA) and differential scanning calorimetry (DSC) under a nitrogen flow. The TGA results show their thermal stability with the 5% weight-reduction temperature (ΔT5%) in the range from ca. 325 °C to 330 °C (Table 1). The DSC traces of the copolymers have revealed their high glass-transition temperatures (Tg) ca. 150 °C (Table 1). The good thermal properties will guarantee their application in OLEDs. It has been taken for granted that the materials with carbon–carbon triple bonds should show disadvantage in the field of EL due to their poor thermal stability. However, it might be case by case, since emitters with carbon–carbon triple bonds have been successfully employed to construct high-performance OLEDs.27 Hence, the good thermal properties associated with these phosphorescent polymers should guarantee their proper application in PHOLEDs.| Polymers | Absorption λabsa (nm) 298 K | Emission λemb (nm) solution at 298 K/film at 298 K/low temperature at 77 K | ΔT5%/Tg (°C) |
|---|---|---|---|
| a Measured in CH2Cl2 at a concentration of 0.02 mg mL−1.b Measured in CH2Cl2 at a concentration of 0.02 mg mL−1. The lifetimes in the parentheses are provided behind the corresponding emission band. The excitation wavelength for the measure was set at 360 nm. | |||
| P-SC-1 | 257, 294, 325, 344, 415 | 424(0.22 ns), 580(1.2 μs), 634/577, 622/458, 520, 577(13.7 μs), 630 | 325/159 |
| P-SC-2 | 256, 294, 324, 343, 415 | 425(0.21 ns), 583(1.2 μs), 635/580, 622/458, 521, 578(13.5 μs), 630 | 327/151 |
| P-SC-3 | 258, 294, 325, 345, 415, 495 | 424(0.18 ns), 585(1.1 μs), 635/580, 622/458, 521, 578(13.8 μs), 630 | 329/152 |
| P-SC-4 | 259, 294, 325, 345, 415, 495 | 424(0.19 ns), 585(1.2 μs), 636/582, 624/458, 522, 578(13.1 μs), 630 | 334/149 |
| P-C | 257, 293, 323, 345 | 420(0.22 ns)/424/456(29.6 μs), 481, 504 | 330/145 |
In the UV-Vis absorption spectra of the phosphorescent polymers (Fig. 1a and Table 1), the strong absorption bands are mainly located before ca. 370 nm, which can be safely assigned to the π–π* transitions of the bicarbazole units with respect to that of their model polymer P-C. Due to the low content of the phosphorescent units in the copolymers, the contribution from the π–π* transitions of the organic ligands chelated with IrIII centers should be in subordinate place. Generally, these phosphorescent polymers show similar UV-Vis absorption spectra to that of the model polymer P-C. However, with increasing of the content of functionalized IrIII phosphorescent unit, the inconspicuous absorption bands after 400 nm are enhanced gradually, which can be assigned to the metal-to-ligand charge transfer states for both singlet (1MLCT) and triplet (3MLCT) from the functionalized phosphorescent units (Fig. 1a and Table 1).
 | ||
| Fig. 1 (a) UV-Vis absorption spectra for the copolymers in CH2Cl2 at 298 K. (b) The photoluminescent (PL) spectra for the copolymers in CH2Cl2 at 298 K. (c) The PL spectra for the copolymer films at 298 K. (d) The PL spectra at 77 K for the copolymers in CH2Cl2. | ||
The photoluminescent (PL) spectra for these polymetallayne-based phosphorescent polymers are also recorded in both solution and film state (Fig. 1b and c and Table 1). In addition, the low-temperature (77 K) PL spectra for the copolymers are also measured (Fig. 1d). In CH2Cl2 solution at 298 K, all the copolymers show major emission band at ca. 580 nm (Fig. 1b and Table 1), which should be induced by the IrIII phosphorescent units in the polymetallayne backbone due to their microsecond-order lifetime (Table 1). The orange phosphorescent band is enhanced with the increasing of the content of the IrIII phosphorescent units. Besides the phosphorescent emission band, there is a high-energy emission at ca. 420 nm in the PL spectra of the copolymer solutions (Fig. 1b and Table 1). According to its nanosecond lifetime (ca. 0.2 ns) together with the PL spectrum of the model polymer P-C (Table 1 and Fig. 1b), the weak high-energy emission band should come from the radiative decay of the singlet states from the bicarbazole-based polymetallayne segments in the copolymers. In the film with thickness of ca. 200 nm, these weak high-energy emission bands cannot even be detected properly (Fig. 1c). Compared with the PL spectra in solution, the phosphorescent intensity of the copolymer films increases from P-SC-1 to P-SC-3 and then decreases (Fig. 1c), which can be attributed to the T–T annihilation effect among the IrIII phosphorescent units in different polymer backbones due to their relatively high content. From the PL spectra in both solution and solid film, it can be concluded that there should be very efficient energy-transfer from the bicarbazole-based polymetallayne segments to the IrIII phosphorescent units, which is quite similar to that involved in traditional phosphorescent polymers with organic conjugated backbones. This result has clearly indicated great potential of polymetallaynes for developing phosphorescent polymers.
Differently, the PL spectra for the solutions of the traditional phosphorescent polymers typically exhibit predominated fluorescent emission bands induced by the singlet states of the pure organic conjugated main chains, and the emission from the phosphorescent moieties is weak or even cannot be detected properly in some cases.28–33 On the contrary, the copolymers P-SC-1–P-SC-4 can show phosphorescence dominated PL spectra even in solution with low content of IrIII phosphorescent moieties (Fig. 1b), indicating the efficient energy-transfer from polymer backbone to the IrIII phosphorescent units. Obviously, if only energy-transfer process from the singlet states of the platinum(II) polymetallayne backbone to the IrIII phosphorescent units involved, the copolymers P-SC-1–P-SC-4 should show similar PL behavior in their solution to that of the traditionally conjugated phosphorescent polymers. Hence, other energy transfer process should be involved in these novel phosphorescent polymers. Based on the structural character of these polymers, the other most likely energy-transfer process in P-SC-1–P-SC-4 should be the triplet of the bicarbazole-based polymetallayne backbone to the functional IrIII phosphorescent units. Clearly, if the triplet energy-transfer process would happen, the lifetime of the phosphorescent bands from these polymers will become longer compared with that of the free phosphorescent monomer M-Ir, since the triplet lifetime of platinum(II) polymetallaynes is typically longer than the IrIII phosphorescent complexes.17–19,34,35 In order to confirm this energy transfer process, the lifetimes (Table 1) and 77 K PL spectra (Fig. 1d) for the phosphorescent polymers, P-C and M-Ir have been obtained. As expected, these phosphorescent polymers do exhibit much longer phosphorescent lifetimes (τp, ca. 1.2 μs at 298 K and ca. 13.0 μs at 77 K) than that of the free monomer M-Ir at both 298 K (ca. 0.51 μs) and 77 K (ca. 4.5 μs). In addition, the model polymer P-C can show strong phosphorescence (ca. 460 nm) at 77 K (Fig. 1d). However, the polymers P-SC-1–P-SC-4 can exhibit the PL spectra with dominated phosphorescent bands from the IrIII phosphorescent units at 77 K despite of their low content and only very weak phosphorescent signals from the polymetallayne backbone can be observed (Fig. 1d). So, all these results have clearly shown the efficient energy transfer process from the triplet of the polymetallayne backbone to the IrIII phosphorescent units in P-SC-1–P-SC-4. Obviously, this triplet energy-transfer process should be absent in the traditional conjugated phosphorescent polymer solutions under photo-excitation, since the pure organic conjugated backbones prefer to generate singlet excited states rather than the triplet ones in photo-excitation process.
Based on the results aforementioned together with the UV-vis absorption and PL spectra for P-C and M-Ir in Fig. 2a, the whole picture about energy transfer processes involved in P-SC-1–P-SC-4 can be figured out. Based on the UV-vis absorption spectrum of the model polymer P-C (Fig. 2a), the 360 nm light can excite the polymetallayne backbones of the phosphorescent polymers to form the first singlet states (S1p). Then, there should be cascade energy-transfer process from S1p to 1MLCTO (singlet states of MLCT in M-Ir), which is converted via inter-system crossing (ISC) into emissive 3MLCTO (triplet states of MLCT in M-Ir) (Fig. 2b) to induce the orange phosphorescence signal in P-SC-1–P-SC-4 (Fig. 1b). This energy-transfer process should be quite similar to that in traditional phosphorescent polymers with organic conjugated backbones. Differently, the platinum(II) ions along the polymetallayne backbones of P-SC-1–P-SC-4 can effectively induce triplet states (T1p) from S1p through ISC process, which have been shown by the strong phosphorescent signal in the PL spectrum of the model polymer P-C at 77 K (Fig. 2a). Owing to the good overlap between the MLCT absorption bands of M-Ir and phosphorescent bands from T1p (Fig. 2a), the energy-transfer from T1p to the emissive 3MLCTO can occur very efficiently and hence induce the strong orange phosphorescent signals in of P-SC-1–P-SC-4 (Fig. 2b). Without the heavy metal ions, the organic conjugated backbones in traditional phosphorescent polymers cannot effectively produce triplet excited states in the photo-excitation process. Hence, this energy-transfer pathway might be absent in the traditional phosphorescent polymers. Additionally, the 360 nm light can also induce excitation of the organic ligands of the IrIII phosphorescent units in P-SC-1–P-SC-4 according to the UV-Vis absorption spectrum of M-Ir (Fig. 2a). There should be energy-transfer process from the singlet of the organic ligands S1O to 1MLCTO, which can be transformed into emissive 3MLCTO via ISC to induce phosphorescence from the IrIII units as well. However, compared with the previous two energy-transfer pathways, this process should be inessential due to the following reasons: (1) owing to the low content of the phosphorescent IrIII units in the polymers, their ligands can only absorb very little energy form the excitation light. (2) Due to the higher energy level of S1O than that of S1p (Fig. 2a), energy-transfer from S1O to S1p can happen to disfavor this process as well (Fig. 2b).
 | ||
| Fig. 2 (a) UV-Vis spectra for P-C and M-Ir together with the PL spectrum of P-C in CH2Cl2 at both 298 K and 77 K. (b) Energy-transfer sketch involved in the orange phosphorescent polymers. | ||
In these phosphorescent copolymers, both electron-rich (bicarbazole) and electron-deficient (sulfone) moieties are included in their main chain. Thus, solvatochromic effect might be involved in their PL spectra. In order to investigate this character of these copolymers, their PL spectra have been recorded in the solvents (toluene, THF and DMF) with different polarity. However, the solvatochromic effect of these copolymers is unobvious, since their phosphorescent maxima red-shifts ca. 8 nm with increasing the polarity of the solvents (Fig. S3 in ESI†). The solvatochromic effect of these copolymers is nearly the same to that of the phosphorescent monomer M-Ir. These results might be ascribed to the fact the Pt(II) ions in the polymer backbone have blocked the interactions between the electron-rich and electron-deficient moieties due to their weak conjugating ability aforementioned.
3.3. Electrochemical properties
The electrochemical behaviors of these phosphorescent polymers have been characterized by the cyclic voltammetry (CV) measurements with ferrocene (Fc) as the standard under nitrogen atmosphere. All the phosphorescent polymers show a quasi-reversible oxidation process at ca. 0.4 V vs. Fc/Fc+ and no detectable reduction procedure has been found (Table 2 and Fig. S4 in ESI†). With the aim to assign the oxidation process, the CV measurement of P-C are also carried out. The model polymer P-C exhibits Ep at ca. 0.33 V, which is quite similar to those of the phosphorescent polymers. Owing to the fact that the platinum(II) centers typically show irreversible oxidation process with Ep at ca. 0.5 V,36 the oxidation peak should be assigned to the bicarbazole units in the backbone of the phosphorescent polymers. Due to their low content in the polymers, the CV signals from the IrIII phosphorescent units cannot be detected properly. The low oxidation potential associated with these phosphorescent polymers indicates their good hole injection abilities. Together with the excellent hole-transporting features of the carbazole moieties,37 these phosphorescent polymers show possess good hole injection/transporting (HI/HT) properties, which will benefit their EL performances.3.4. Electroluminescent characterization
As aforementioned, the efficient energy-transfer from the triplet sates of the backbone to the IrIII phosphorescent units under photo-excitation can furnish strong phosphorescence to the solution of these novel phosphorescent polymers with polymetallayne backbones. Even in the PHOLEDs based on traditional conjugated phosphorescent polymers, there is also predominant energy-transfer process from the triplet sates of the conjugated backbones to the phosphorescent units in the EL process due to the fact that the majority of the generated excitons associated with the pure organic conjugated backbones in electric excitation is in triplet state. Thus, the energy-transfer between triplet states is very crucial to the PHOLEDs based on phosphorescent polymers. Clearly, facilitating the formation of triplet states by the platinum(II) ions along the polymetallayne backbones in P-SC-1–P-SC-4 should benefit their EL process. Additionally, due to the fact that platinum(II) ions show weak ability in extending conjugation for the much weaker dπ–pπ conjugation effect,17–19,34,35 the polymetallayne backbones of P-SC-1–P-SC-4 should show high triplet energy-level (ca. 2.73 eV) confirmed by the PL spectrum of P-C at 77 K (Fig. 1d). Thus, the novel polymetallayne backbones can effectively block the reverse energy-transfer process. Furthermore, the functionalized IrIII phosphorescent units showing EI/ET features can balance the HI/HT ability associated with the bi-carbazole-based polymetallayne backbones of P-SC-1–P-SC-4, which should benefit the EL performances of these phosphorescent polymers as well. With all the features, these novel phosphorescent polymers should show great potential in PHOLEDs. Hence, their EL potentials have been characterized as well.The solution-processed PHOLEDs based on these polymers have be constructed with the structure of ITO/PEDOT:PSS (45 nm)/Emission layer, EML (35 nm)/TPBi (45 nm)/LiF (1 nm)/Al (100 nm) (Fig. 3). The PEDOT:PSS layer acts as hole-injection layer (HIL). The 1,3,5-tris(1-phenyl-1H-benzo[d]imidazol-2-yl)benzene (TPBi) layer shows the function of both hole-blocking and electron-transporting, while LiF serves as an electron-injection layer. When proper voltage was applied to the PHOLEDs, intense orange electrophosphorescence at ca. 580 nm can be observed (Fig. 4). For devices O1–O4 with P-SC-1–P-SC-4 as emitter (Fig. 3), they show orange EL representing similar line-shape to that of corresponding polymers in solid film, indicating the origin of EL is from the phosphorescent units. The slight red-shift effect in the EL spectrum of device O4 should be induced by the aggregation among the phosphorescent units in different polymer backbones. No detectable EL bands from the polymetallayne backbones of the polymers have been found, indicating the complete energy-transfer in the EL processes (Fig. 4). This result should benefit from both the high triplet energy-level of the bicarbazole-based polymetallayne backbones and the highly efficient triplet energy-transfer process aforementioned (Fig. 2b), indicating the crucial role played by the polymetallayne backbones in the EL process of the concerned phosphorescent polymers.
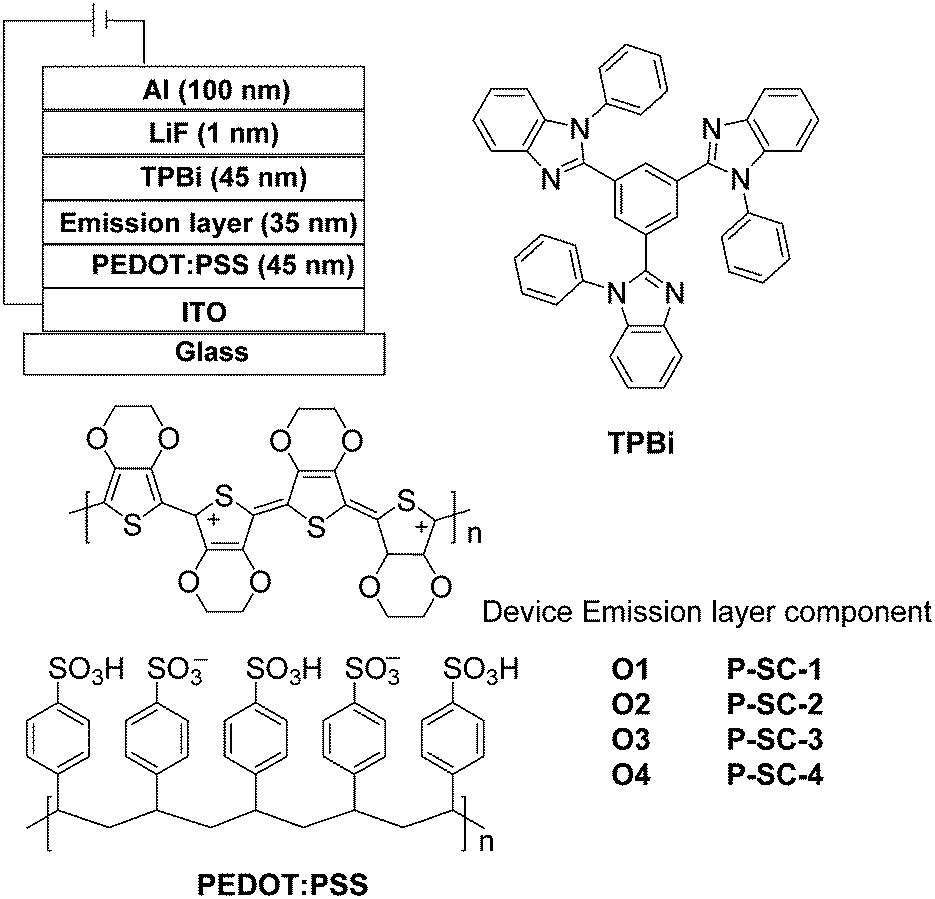 | ||
| Fig. 3 The configuration of the PHOLEDs made from the orange phosphorescent polymers and the chemical structures for the involved functional materials. | ||
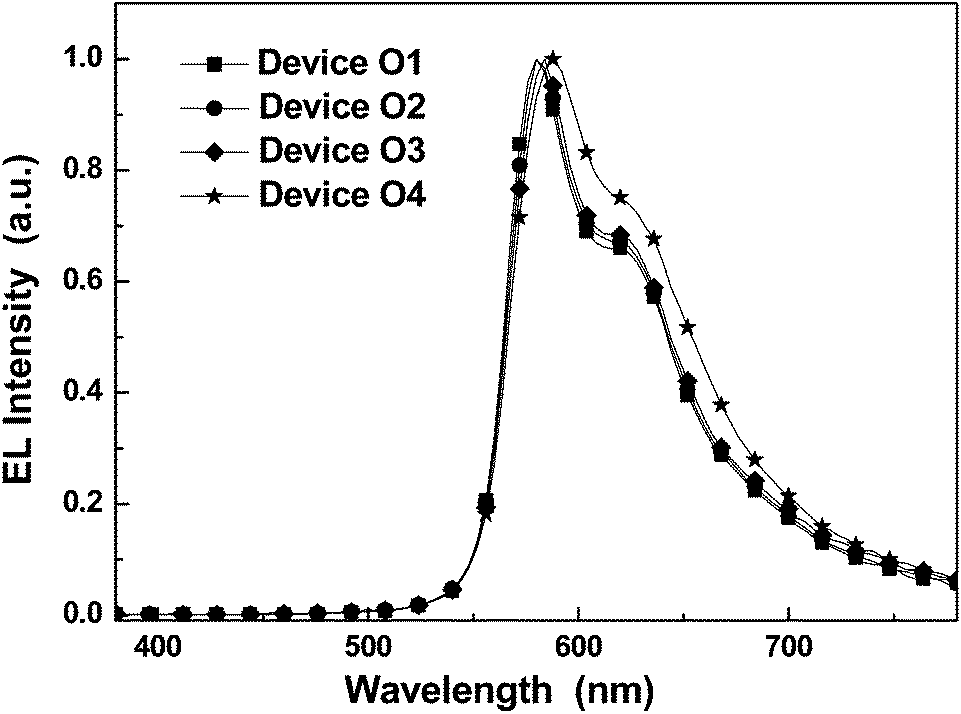 | ||
| Fig. 4 The EL spectra for the solution-processed PHOLEDs at ca. 12 V. | ||
The current-density–voltage–luminance (J–V–L) curves for the PHOLEDs are shown in Fig. 5a. The corresponding EL data are summarized in Table 3. All the devices show the turn-on voltages in the range from 4.4 V to 5.4 V (Table 3). Among all the PHOLEDs, device O3 can furnish the best EL performances with peak luminance (Lmax) of 10369 cd m−2 at 16.0 V, current efficiency (ηL) of 9.17 cd A−1, external quantum efficiency (ηext) of 4.50% and power efficiency (ηP) of 4.04 lm W−1 (Fig. 5b and Table 3). In addition, device O2 can also show nice peak EL performance with Lmax of 6477 cd m−2 at 18.7 V, ηL of 7.50 cd A−1, ηext of 3.79% and ηP of 3.59 lm W−1 (Table 3 and Fig. S5 in ESI†). Despite of their inferior EL efficiencies compared with those from device O2 and O3, the device O1 and O4 still bring ηL higher than of 3.6 cd A−1 (Table 3 and Fig. S5 in ESI†). Obviously, the nice EL performances associated with these orange phosphorescent polymers have indicated the great potential of the platinum(II) polymetallayne backbones in developing novel phosphorescent polymers, which has been rarely evaluated in the field of PHOLEDs. In addition, the contribution to the high EL efficiencies from the functionalized phosphorescent IrIII units cannot be totally excluded, since they can promote the EI/ET process in the devices indicated by the enhancing of the current density in the electron-only devices for P-SC-1–P-SC-4 with the increasing of content for the IrIII units (Fig. S6 in ESI†). Compared with the analogs emitting primary colors, the yellow/orange phosphorescent polymers are in limited number. However, the yellow/orange phosphorescent polymers can show their potential in achieving white OLEDs through complementary color strategy. Some functionalized yellow/orange phosphorescent polymers can achieve ηL of 10.4 cd A−1 or even 30.54 cd A−1 due to their functional ambipolar moieties in the backbones.38,33 However, some conjugated orange phosphorescent polymers with oxadiazole moieties exhibit ηL of 0.61 cd A−1.39 The fluorescent OLEDs based on green-emitting bithiozole-based platinum(II) polymetallaynes just show ηL of 0.11 cd A−1.16 Despite that some other highly efficient phosphorescent polymers has been reported,38,40–46 most of them typically show ηL less than 5.0 cd A−1.47,48 Hence, compared with the general EL performances of the phosphorescent polymers, these novel orange phosphorescent polymers definitely can show very attractive EL performances, indicating the great potential of the novel polymer skeleton for achieving highly efficient phosphorescent polymers. The obtained EL data are just preliminary results. We are sure that much better EL performances can be expected through further polishing the polymer structure as well as device optimization. Hence, the concerned results can provide very valuable information for design and synthesis of highly efficient phosphorescent polymers.
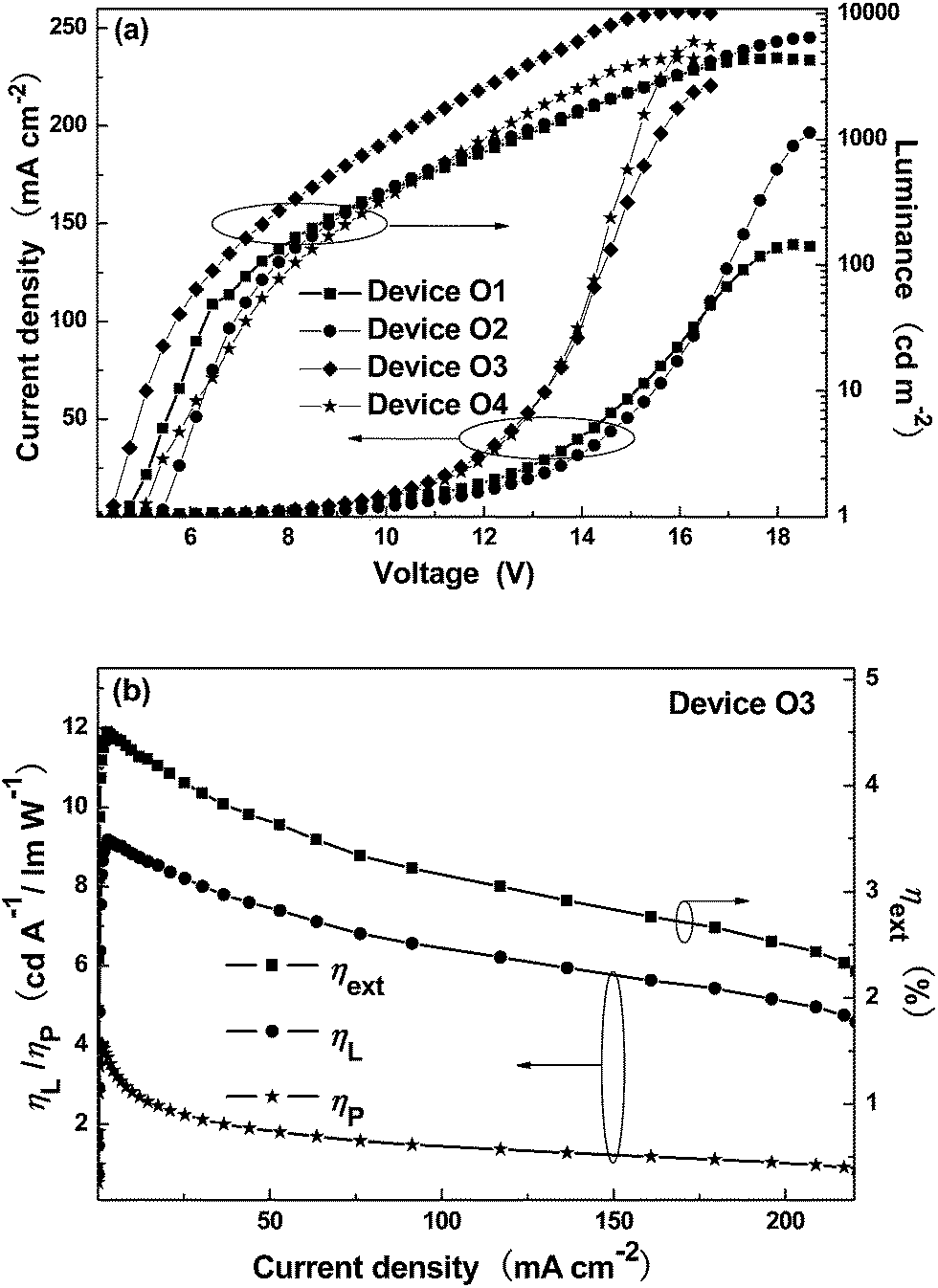 | ||
| Fig. 5 (a) The current-density–voltage–luminance (J–V–L) curves for all the devices. (b) The relationship between EL efficiencies and current density for device O3. | ||
| Device | Polymers | Vturn-on (V) | Luminance Lmaxa (cd m−2) | ηext (%) | ηL (cd A−1) | ηp (lm W−1) | λmaxd (nm) |
|---|---|---|---|---|---|---|---|
| a Maximum values of the devices. Values in parentheses are the voltages at which they were obtained.b Values were collected at 100 cd cm−2.c Values collected at 1000 cd cm−2.d Values were collected at 12 V and CIE coordinates (x, y) are shown in parentheses. | |||||||
| O1 | P-SC-1 | 4.8 | 4460 (17.9) | 2.36 (9.51)a | 5.00 (9.51) | 1.82 (8.15) | 580 |
| 1.95b | 4.16 | 1.77 | (0.57, 0.43) | ||||
| 2.03c | 4.35 | 1.03 | |||||
| O2 | P-SC-2 | 5.4 | 6477 (18.7) | 3.79 (11.82) | 7.50 (9.85) | 3.59 (9.85) | 580 |
| 2.86 | 6.13 | 2.39 | (0.57, 0.43) | ||||
| 3.05 | 6.41 | 1.63 | |||||
| O3 | P-SC-3 | 4.4 | 10369 (16.0) |
4.50 (7.81) | 9.17 (7.81) | 4.04 (6.46) | 584 |
| 4.20 | 8.45 | 4.01 | (0.57, 0.42) | ||||
| 4.30 | 8.75 | 2.72 | |||||
| O4 | P-SC-4 | 5.1 | 4445 (15.9) | 1.88 (9.85) | 3.63 (9.85) | 1.30 (8.15) | 588 |
| 1.73 | 3.28 | 1.29 | (0.58, 0.42) | ||||
| 1.75 | 3.39 | 0.89 | |||||
4. Conclusion
Novel orange phosphorescent polymers have been successfully developed based on both bicarbazole platinum(II) polymetallayne backbones and functionalized phosphorescent IrIII units with EI/ET characters. These phosphorescent polymers not only show high triplet energy-level backbone to block undesired reverse energy-transfer process, but also fulfill highly efficient triplet energy-transfer from the polymetallayne backbone to the IrIII phosphorescent units. Due to these merits, highly efficient orange solution-processed PHOLEDs can be made from these novel phosphorescent polymers with ηL of 9.17 cd A−1, ηext of 4.50% and ηP of 4.04 lm W−1. The preliminary work should present important inspirations for constructing highly efficient phosphorescent polymers.Acknowledgements
This work was financially supported by Tengfei Project from Xi'an Jiaotong University, the Fundamental Research Funds for the Central Universities, the Program for New Century Excellent Talents in University, the Ministry of Education of China (NECT-09-0651), the Key Creative Scientific Research Team in Shaanxi Province (2013KCT-05), the China Postdoctoral Science Foundation (Grant no. 20130201110034), and the National Natural Science Foundation of China (no. 20902072). The financial supporting from State Key Laboratory for Mechanical Behavior of Materials is also acknowledged.References
- S. J. Liu, Q. Zhao, Y. Deng, Y. J. Xia, J. Lin, Q. L. Fan, L. H. Wang and W. Huang, J. Phys. Chem. C, 2007, 111, 1166–1175 CAS.
- H. Y. Zhen, C. Luo, W. Yang, W. Y. Song, B. Du, J. X. Jiang, C. Y. Jiang, Y. Zhang and Y. Cao, Macromolecules, 2006, 39, 1693–1700 CrossRef CAS.
- G. L. Schulz, X. W. Chen, S. A. Chen and S. Holdcroft, Macromolecules, 2006, 39, 9157–9165 CrossRef CAS.
- K. Zhang, Z. Chen, Y. Zou, C. L. Yang, J. G. Qin and Y. Cao, Organometallics, 2007, 26, 3699–3707 CrossRef CAS.
- A. J. Sandee, C. K. Williams, N. R. Evans, J. E. Davies, C. E. Boothby, A. Kohler, R. H. Friend and A. B. Holmes, J. Am. Chem. Soc., 2004, 126, 7041–7048 CrossRef CAS PubMed.
- J. X. Jiang, Y. H. Xu, W. Yang, R. Guan, Z. Q. Liu, H. Y. Zhen and Y. Cao, Adv. Mater., 2006, 18, 1769–1773 CrossRef CAS.
- Z. H. Ma, J. Q. Ding, B. H. Zhang, C. Y. Mei, Y. X. Cheng, Z. Y. Xie, L. X. Wang, X. B. Jing and F. S. Wang, Adv. Funct. Mater., 2010, 20, 138–146 CrossRef CAS.
- D. A. Poulsen, B. J. Kim, B. Ma, C. S. Zonte and J. M. J. Frechet, Adv. Mater., 2010, 22, 77–82 CrossRef CAS PubMed.
- S. Y. Shao, J. Q. Ding, L. X. Wang, X. B. Jing and F. S. Wang, J. Mater. Chem., 2012, 22, 24848–24855 RSC.
- S. Y. Shao, J. Q. Ding, L. X. Wang, X. B. Jing and F. S. Wang, J. Am. Chem. Soc., 2012, 134, 20290–20293 CrossRef CAS PubMed.
- Z. H. Ma, L. C. Chen, J. Q. Ding, L. X. Wang, X. B. Jing and F. S. Wang, Adv. Mater., 2011, 23, 3726–3729 CrossRef CAS PubMed.
- G. J. Zhou, W. Y. Wong, S. Y. Poon, C. Ye and Z. Y. Lin, Adv. Funct. Mater., 2009, 19, 531–544 CrossRef CAS.
- G. J. Zhou and W. Y. Wong, Chem. Soc. Rev., 2011, 40, 2541–2566 RSC.
- W. Y. Wong, X. Z. Wang, Z. He, K. K. Chan, A. B. Djurisic, K. Y. Cheung, C. T. Yip, A. M. C. Ng, Y. Y. Xi, C. S. K. Mak and W. K. Chan, J. Am. Chem. Soc., 2007, 129, 14372–14380 CrossRef CAS PubMed.
- W. Y. Wong, X. Z. Wang, Z. He, A. B. Djurisic, C. T. Yip, K. Y. Cheung, H. Wang, C. S. K. Mak and W. K. Chan, Nat. Mater., 2007, 6, 521–527 CrossRef CAS PubMed.
- W. Y. Wong, G. J. Zhou, Z. He, K. Y. Cheung, A. M. C. Ng, A. B. Djurisic and W. K. Chan, Macromol. Chem. Phys., 2008, 209, 1319–1332 CrossRef CAS.
- W.-Y. Wong and C.-L. Ho, Coord. Chem. Rev., 2006, 250, 2627–2690 CrossRef CAS PubMed.
- G. J. Zhou, W. Y. Wong, C. Ye and Z. Y. Lin, Adv. Funct. Mater., 2007, 17, 963–975 CrossRef.
- G. J. Zhou, W. Y. Wong, Z. Y. Lin and C. Ye, Angew. Chem., Int. Ed., 2006, 45, 6189–6193 CrossRef CAS PubMed.
- M. S. Khan, M. R. A. Al-Mandhary, M. K. Al-Suti, A. K. Hisahm, P. R. Raithby, B. Ahrens, M. F. Mahon, L. Male, E. A. Marseglia, E. Tedesco, R. H. Friend, A. Kohler, N. Feeder and S. J. Teat, J. Chem. Soc., Dalton Trans., 2002, 1358–1368 RSC.
- Y. You and S. Y. Park, Dalton Trans., 2009, 1267–1282 RSC.
- G. J. Zhou, W. Y. Wong and X. L. Yang, Chem.–Asian J., 2011, 6, 1706–1727 CrossRef CAS PubMed.
- C. Fan and C. Yang, Chem. Soc. Rev., 2014, 43, 6439–6469 RSC.
- G. J. Zhou, X. L. Yang, W. Y. Wong, Q. Wang, S. Suo, D. G. Ma, J. K. Feng and L. X. Wang, ChemPhysChem, 2011, 12, 2836–2843 CrossRef CAS PubMed.
- G. J. Zhou, Q. Wang, C. L. Ho, W. Y. Wong, D. G. Ma, L. X. Wang and Z. Y. Lin, Chem.–Asian J., 2008, 3, 1830–1841 CrossRef CAS PubMed.
- G. J. Zhou, C. L. Ho, W. Y. Wong, Q. Wang, D. G. Ma, L. X. Wang, Z. Y. Lin, T. B. Marder and A. Beeby, Adv. Funct. Mater., 2008, 18, 499–511 CrossRef CAS.
- T. Peng, G. F. Li, Y. Liu, Y. Wu, K. Q. Ye, D. D. Yao, Y. Yuan, Z. M. Hou and Y. Wang, Org. Electron., 2011, 12, 1068–1072 CrossRef CAS PubMed.
- J. Jiang, C. Jiang, W. Yang, H. Zhen, F. Huang and Y. Cao, Macromolecules, 2005, 38, 4072–4080 CrossRef CAS.
- X.-H. Yang, F.-I. Wu, D. Neher, C.-H. Chien and C.-F. Shu, Chem. Mater., 2008, 20, 1629–1635 CrossRef CAS.
- T. Fei, G. Cheng, D. Hu, W. Dong, P. Lu and Y. Ma, J. Polym. Sci., Part A: Polym. Chem., 2010, 48, 1859–1865 CrossRef CAS.
- K. Zhang, Z. Chen, Y. Zou, S. Gong, C. Yang, J. Qin and Y. Cao, Chem. Mater., 2009, 21, 3306–3314 CrossRef CAS.
- H. Tan, J. Yu, K. Nie, J. Chen, X. Deng, Z. Zhang, G. Lei, M. Zhu, Y. Liu and W. Zhu, Polymer, 2011, 52, 4792–4797 CrossRef CAS PubMed.
- M. Lian, Y. Yu, J. Zhao, Z. Huang, X. Yang, G. Zhou, Z. Wu and D. Wang, J. Mater. Chem. C, 2014, 2, 9523–9535 RSC.
- S. J. Davies, B. F. Johnson, M. S. Khan and J. Lewis, J. Chem. Soc., Chem. Commun., 1991, 187–188 RSC.
- N. J. Long and C. K. Williams, Angew. Chem., Int. Ed., 2003, 42, 2586–2617 CrossRef CAS PubMed.
- P.-I. Kvam, M. Puzyk, K. P. Balashev and J. Songstad, Acta Chem. Scand., 1995, 49, 335–343 CrossRef CAS PubMed.
- J. Ding, B. Zhang, J. Lü, Z. Xie, L. Wang, X. Jing and F. Wang, Adv. Mater., 2009, 21, 4983–4986 CrossRef CAS PubMed.
- S. Shao, J. Ding, L. Wang, X. Jing and F. Wang, J. Mater. Chem., 2012, 22, 24848–24855 RSC.
- W. Zhang, H. Jin, F. Zhou, Z. Shen, D. Zou and X. Fan, J. Polym. Sci., Part A: Polym. Chem., 2012, 50, 3895–3903 CrossRef CAS.
- J. H. Park, T.-W. Koh, J. Chung, S. H. Park, M. Eo, Y. Do, S. Yoo and M. H. Lee, Macromolecules, 2013, 46, 674–682 CrossRef CAS.
- W.-Y. Lai, J. W. Levell, M. N. Balfour, P. L. Burn, S.-C. Lo and I. D. Samuel, Polym. Chem., 2012, 3, 734–740 RSC.
- Z. Ma, L. Chen, J. Ding, L. Wang, X. Jing and F. Wang, Adv. Mater., 2011, 23, 3726–3729 CrossRef CAS PubMed.
- F. I. Wu, X. H. Yang, D. Neher, R. Dodda, Y. H. Tseng and C. F. Shu, Adv. Funct. Mater., 2007, 17, 1085–1092 CrossRef CAS.
- C. H. Chien, S. F. Liao, C. H. Wu, C. F. Shu, S. Y. Chang, Y. Chi, P. T. Chou and C. H. Lai, Adv. Funct. Mater., 2008, 18, 1430–1439 CrossRef CAS.
- S. Shao, J. Ding, T. Te, Z. Xie, L. Wang, X. Jing and F. Wang, Adv. Mater., 2011, 23, 3570–3574 CrossRef CAS PubMed.
- S. Shao, J. Ding, L. Wang, X. Jing and F. Wang, J. Am. Chem. Soc., 2012, 134, 15189–15192 CrossRef CAS PubMed.
- S. Gong, C. Yang and J. Qin, Chem. Soc. Rev., 2012, 41, 4797–4807 RSC.
- G. Zucchi, D. Tondelier, Y. Bonnassieux and B. Geffroy, Polym. Int., 2014, 63, 1368–1377 CrossRef CAS.
Footnote |
| † Electronic supplementary information (ESI) available: Synthetic detail for some organic compounds, PL spectra for the polymer solution, EL spectra for device O1–O4, and some electroluminescent data for the concerned devices. See DOI: 10.1039/c4ra16286b |
| This journal is © The Royal Society of Chemistry 2015 |