
Activation of giant silicalite-1 monocrystals combining rapid thermal processing and ozone calcination
Marta Navarroab,
Ester Mateoc,
Beatriz Diosdadod,
Michael Tsapatsise and
Joaquín Coronas*a
aChemical and Environmental Engineering Department and Nanoscience Institute of Aragón (INA), Universidad de Zaragoza, 50018 Zaragoza, Spain. E-mail: coronas@unizar.es; Fax: +34 976 761879; Tel: +34 976 762471
bDepartment of Earth Sciences, Universidad de Zaragoza, 50009 Zaragoza, Spain
cDepartamento de Didáctica de las Ciencias Experimentales, Universidad de Zaragoza, 50009 Zaragoza, Spain
dX-Ray Diffraction and Fluorescence Analysis Service, Universidad de Zaragoza, 50009 Zaragoza, Spain
eDepartment of Chemical Engineering and Materials Science, University of Minnesota, Minneapolis, MN 55455, USA
First published on 4th February 2015
Abstract
A rapid thermal processing (RTP) technique combined with ozone/oxygen calcination was applied to activate and heal the structure of a 1.1 mm2 section of a silicalite-1 giant monocrystal. Crystallinity was monitored by single crystal X-ray diffraction and high sensitive thermogravimetry (TGA) was performed to prove the complete removal of the structure directing agent.
Introduction
Silicalite-1, a pure silica zeolite with an MFI type structure, has attracted considerable attention due to its particular framework. It contains two intersecting channels, one sinusoidal and parallel to (100) with elliptical cross sections (0.51 × 0.55 nm in size), and the other straight, circular in section (0.53 × 0.56 nm) and parallel to (010).1 This particular channel system plays an important role in diffusion processes,2 membrane permeation properties3 and even crystal growth habit.4 The orientation or preferential growth of the MFI type zeolite crystals along the b axis, i.e. the (010) direction, promotes a better performance in membrane separations, increasing the flux and selectivity to specific molecules.3 Large silicalite-1 monocrystals could be sectioned perpendicularly to the b axis, yielding to lamellae free of intergrowth defects. With the advantage of porosity-morphology control, such lamellae, as zeolites or microporous materials in general,5,6 could serve as effective self-supported membranes,7 fillers in mixed matrix membranes,8 sensors9 and other microdevices.10For such samples to be accessible for guest molecules of interest and useful as separation membranes,6 it is important to remove the occluded template or organic structure directing agent (OSDA) molecules while retaining the inorganic framework.11 Tetrapropylammonium hydroxide (TPAOH) has been used as an OSDA, although other organic templates can be used to synthesize large silicalite-1 crystals.12 During calcination at high temperatures, mechanical tensions are produced in the zeolite framework and in consequence cracks usually arise; in addition, carbon residues are formed along the diffusion paths. All these issues become crucial when the performance of large crystals is considered. Herein we demonstrate how a rapid thermal processing (RTP)13 technique heals the structure of up to a 1.1 mm2 section of silicalite-1 giant monocrystal. Furthermore, oxygen/ozone mixtures were used for the complete removal of the OSDA under mild temperature conditions (350 °C). Single crystal diffraction was applied to study the effect of combining both methodologies in the zeolite structure,12,14 and high sensitive thermogravimetry (TGA) able to register the weight loss of a single crystal (ca. 300 μg) was performed to confirm OSDA removal. Finally, even though both RTP treatment and ozone calcination have previously been reported,13–15 this is the first time that OSDA was completely removed from giant silicalite-1 crystals providing evidence of structure and crystallinity preservation.
Experimental
Synthesis and sectioning of giant silicalite-1 crystals
Synthesis of giant zeolite crystals was carried out following the methodology already published.12 This method is based on slowing down the dissolution of the silica source using low surface area materials of silicon, such as two quartz glass tubes, following the so-called BMD (“bulk material dissolution”) technique.16 Prior to the addition of reactants, a thorough cleaning process was achieved in the Teflon liners and silica tubes. Then, 25.35 g of tetrapropylammonium hydroxide solution (TPAOH 1 M, Aldrich) was used as the structure directing agent and afterwards 1.68 g of HF (48 wt% in water, Aldrich) was added as a nucleation suppressing agent. The molar composition of the gel was 24TPAOH![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) :38HF:1000H2O, and the hydrothermal synthesis was carried out at 200 °C for 10 days. Crystals were recovered by handpicking, washed thoroughly with distilled water and dried at 70 °C overnight.
:38HF:1000H2O, and the hydrothermal synthesis was carried out at 200 °C for 10 days. Crystals were recovered by handpicking, washed thoroughly with distilled water and dried at 70 °C overnight.
For sectioning the silicalite-1 crystals, a vertical diamond wire saw (Well, 3242 model) was used, equipped with two micrometers for accurate positioning of the sample and for setting the depth of a slice. The crystal was mounted on an alumina support in the desired position and glued with Quickstick 135 mounting wax to avoid any deviation while cutting.
Calcination treatments
For RTP treatment, silicalite-1 samples were placed over a quartz reactor to heat them up to 700 °C within 25 s, using an infrared heating chamber (Research Inc., model E4-10), supplied with a cabinet temperature controller (model 915). The maximum temperature was held for 2 min, and then the sample was cooled down using a water cooling system (Research Inc., model C100-1). The thermal treatment was performed in oxygen atmosphere (1.2 L min−1).For ozone calcination, a single crystal is placed in a quartz reactor and calcined in a cross-flow system using an O2/O3 mixture, feeding O2 (20 mL min−1) through a O3 generator (Neosyt NEO 500), giving an ozone production of approximately 0.3 mL min−1 (1.5%). The temperature was maintained at 350 °C for 12 h with heating and cooling rates of 0.2 and 1 °C min−1, respectively. Pure oxygen calcination was not carried out due to crack formation.14 The effect of calcination conditions and gas atmospheres used on template removal was studied by using a TA Instruments Thermogravimetric Analyzer TGA-Q5000 IR with a resolution of 0.01 μg. With this equipment, calcination analyses were carried out in air (50 mL (STP) min−1) at 600 °C for 8 h, with heating and cooling rates of 0.5 and 1 °C min−1, respectively.
Characterization
Optical microscopy (Olympus Bx41 TRF) was routinely employed to observe, measure and classify the handpicked specimens. Single crystal X-ray data were adquired on a Xcalibur, Sapphire-3 diffractometer equipped with a graphite monochromator (Moκα radiation, λ = 0.71073 Å) and analysed using the CrysAlisPro package. [CrysAlisPro, Agilent Technologies, Version 1.171.37.31 (release 14-01-2014 CrysAlis171.NET) (compiled Jan 14 2014, 18:38:05)]. Data collection commences indexing the spots from several image frames. The software automatically does “peak hunting” and finds a unit cell. The same variables (default settings) and background are used for analysing every sample. Orthogonal projections of the Ewald sphere were presented from the original unit cell calculated by the software. Then, monocrystallinity percentages were obtained comparing both the unit cell defined for silicalite-1 (ref. 17) and that calculated by the software.Morphology, size and quality of the samples were determined with scanning electron microscopy (SEM) using an Inspect F50 field emission gun scanning electron microscope, operated at 5 and 10 kV. 3D images of the silicalite-1 slice were obtained with a SENSOFAR PLμ 2300 confocal microscope, operating in reflection mode.
Results and discussion
Recently, we have reported the synthesis and characterization of giant silicalite-1 monocrystals of different sizes and aspect ratios by single crystal X-ray diffraction (XRD).12,14 The crystals were prepared applying the “bulk material dissolution” (BMD) technique.16 Cross sections of silicalite-1 monocrystals larger than 1 mm in size were obtained along the b axis using a diamond saw, showing minor roughness and no cracks on their surface (Fig. 1). Even though the cutting process must be optimized by a method allowing ultrathin lamellae to be obtained, the results are promising since we were able to substantially reduce the thickness of the sample from 610 to 160 μm of Lb (where Li is the dimension along i axis). Moreover, our sections did not show evident cracks after the process, as can be observed from the confocal profilometry imaging (Fig. 1c). We will demonstrate that the cutting process did not affect the bulk monocrystallinity of the sample. | ||
| Fig. 1 Silicalite-1 cross section along (010) axis: (a) outline of a slice along b axis in a typical coffin shape crystal; (b) obtained slice of 160 μm in thickness; (c) confocal profilometry image of (b) slightly deviated from the medium b axis length. | ||
As determined by single crystal XRD, as-synthesized silicalite-1 crystals greater than 250 μm in size tend to show a relatively high density of structural defects and fractures, especially after calcination at high temperatures.12 The silicalite-1 crystal being the object of our study (La = 990 μm; Lb = 610 μm; Lc = 1300 μm; i.e. with a section along the (010) axis of ca. 1.3 mm2) showed these features under optical microscopy also before calcination (Fig. 2a). Furthermore, single crystal XRD (Fig. 3a) revealed the presence of polycrystalline domains and microstructural defects, represented by spots (reflections) outside their expected positions from the silicalite-1 orthorhombic unit cell.17 In detail, only 47.0% of the analyzed data fitted with the orthorhombic cell calculated for this silicalite-1 crystal (Table 1). After laminating the crystal, its thickness was reduced from 610 μm to 160 μm and its section along (010) to 1.1 mm2 (due to the fact that the crystal thickness was not constant) and the fraction of the as-made crystal that presented a greater amount of defects (those present in the growing external surface) was removed. In consequence, the monocrystallinity of the subsequent lamella increased, the percentage of the analyzed data that fitted with the unit cell being 56.0%.
 | ||
| Fig. 2 (a and b) Transmitted light optical microscopy images of silicalite-1 sample: (a) as-synthesized crystal (La = 990 μm; Lb = 610 μm; Lc = 1300 μm); (b) cross section along b axis (La = 990 μm; Lb = 160 μm; Lc = 1100 μm) after RTP treatment. Horizontal bars indicate 1000 μm. | ||
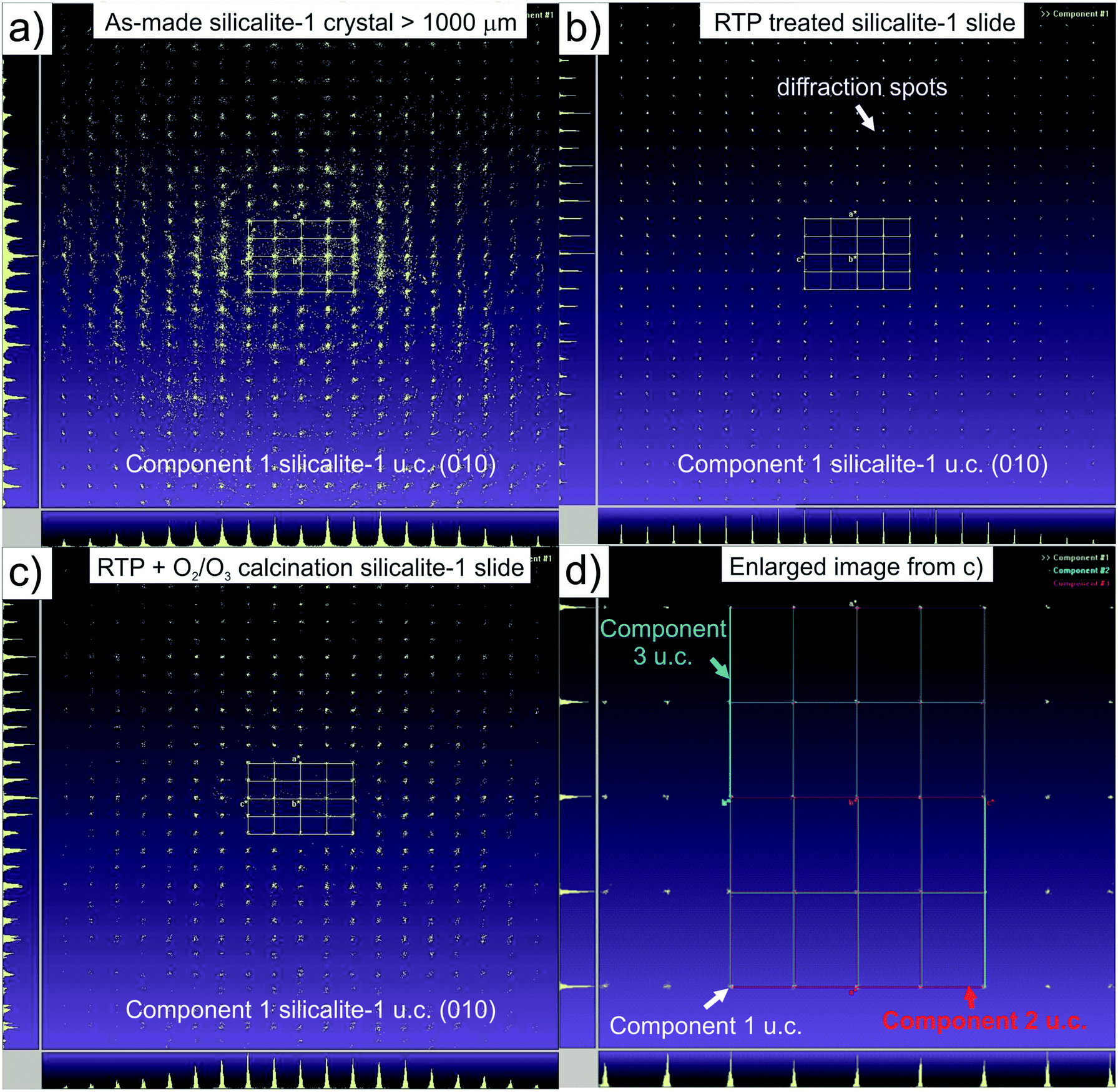 | ||
| Fig. 3 Orthogonal projection of the Ewald sphere of silicalite-1 samples along (010) direction: (a) component 1 of the as-made crystal (La = 990 μm; Lb = 610 μm; Lc = 1300 μm) unit cell; (b–d) silicalite-1 slice (La = 990 μm; Lb = 160 μm; Lc = 1100 μm): (b) component 1 of the unit cell after RTP; (c) component 1 of the unit cell after RTP treatment and ozone calcination; (d) detail of the projection in (c) with the three unit cell components drawn. | ||
| Silicalite-1 sample | Monocrystallinity | Change in monocrystallinity |
|---|---|---|
| a Sample treated by RTP (700 °C for 2 min).b Sample treated by RTP (700 °C for 2 min) and calcined using oxygen/ozone atmosphere (350 °C for 12 h). Monocrystallinity percentages were calculated comparing both the unit cell defined for silicalite-1 (ref. 17) and that calculated by the software using up to 40000 spots from several image frames. |
||
| As-made crystal | 47.0% | — |
| Slice | 56.0% | +9% |
| RTP treated slicea | 99.6% | +43.6% |
| RTP + O2/O3 calcination sliceb | 89.7% | −9.9% |
As reported by Choi et al.,13 a sudden RTP treatment under an infrared lamp at 700 °C during 2 min diminished the formation of grain boundary defects in zeolite samples by the condensation of terminal silanol groups (Si–OH), thus preventing fracture formation. To further study the RTP treatment effect on the zeolite microstructure, this large silicalite-1 cross section along the b axis was analyzed again by single crystal XRD. In Fig. 3b, an orthorhombic unit cell diagram is drawn over the reflections found along the b direction. We can observe how intense and well defined these peaks are in comparison with the intensity diagram obtained from the original crystal in Fig. 3a. In fact, after RTP treatment (700 °C, 2 min) almost all the reflections matched the calculated unit cell (99.6%, Table 1) in comparison with the 56.0% matching registered after the sectioning. These findings suggest that RTP treatment healed the structure of silicalite-1 crystals up to a point that samples can be considered as practically monocrystalline after being submitted to this process. Moreover, the 160 μm silicalite-1 slice showed a more transparent surface under optical microscopy, although the superficial crack was still present from the growing step of the original crystal (Fig. 2b).
Single-step18 or two-step13 RTP processes have been used before to remove completely the template occluded in the zeolite pores, depending on the thickness of the polycrystalline sample. Using high sensitivity TGA equipment, air atmosphere and 600 °C as the maximum temperature, the weight loss of a single crystal greater than 1000 μm in size treated with RTP was registered (Fig. 4). This sample (La = 470 μm; Lb = 460 μm; Lc = 1050 μm) weighed 339.2 μg and suffered a 13.0% weight loss at 370 °C. It should be noted that this weight loss is higher (0.8%) than that of the as-made crystal (311.4 μg) which lost 12.2% of its weight at 374 °C under the same experimental conditions. This result is in agreement with that already published for crystals greater than 1000 μm (12.3%).12 After RTP followed by conventional air calcination (RTP alone cannot completely remove the OSDA as it was already proved) at 600 °C for 8 h,13 the crystal integrity seemed to be better preserved in comparison with other samples calcined without previous RTP (Fig. 5a),12,14 but numerous superficial cracks and carbon deposits were inferred by optical microscopy images (not shown). After the combination of both processes, single crystal XRD (Fig. 5b) showed that only 36.1% of all the measured reflections fitted the calculated orthorhombic unit cell. Minor percentages (18.8 and 14.8%) also fitted the calculated unit cell, but corresponding to different components slightly rotated (0.07° and 2.5°) with respect to the main component (Fig. 5c). The remaining reflections measured (30.3%) corresponded to highly defective areas inside the crystal and could not be fitted to other unit cell components. These features reveal the severe decrease in monocrystallinity in the sample after the application of drastic calcination conditions even if RTP treatment is applied in advance.
 | ||
| Fig. 4 Weight loss curves under air atmosphere for silicalite-1 monocrystalline samples with Lc > 1000 μm in size: as-made and RTP treated crystal and RTP-ozone treated slice. | ||
 | ||
| Fig. 5 (a) SEM image of a single silicalite-1 crystal after RTP treatment and conventional calcination; (b and c) orthogonal projection of the Ewald sphere along (100) direction of a silicalite-1 crystal RTP treated and conventionally calcined: (b) reflections that do not fit with the orthorhombic unit cell of the component 1 drawn in the image; (c) enlargement of the slightly rotated orthorhombic unit cell components after fitting all the spots measured. | ||
Ozone, already proved as a suitable gas for removing the organic template from silicalite-1 large crystals and membranes while preserving their structure,14,19 was assayed as the appropriate atmosphere to calcine samples under mild temperature conditions after RTP treatment. The maximum calcination temperature was set at 350 °C for 12 h with very slow heating/cooling rates (0.2/1 °C min−1). Once again, high sensitivity TGA equipment was used to prove the effectiveness of ozone calcination in the removal of the organic template, in combination with RTP treatment (Fig. 4). The initial weight of the millimetric silicalite-1 cross section was 480.0 μg. After an initial weight loss of around 100–150 °C (1.2%) attributed to occluded water, the sample underwent a small weight loss (2.0%) assigned to remaining carbon deposits, always difficult to eliminate from large silicalite-1 crystals,14,20 observed in the surface of the sample that shows brownish color. This weight loss value indicates that template removal was achieved in a high yield from our sample by the combination of both methodologies, which preserved the zeolite structure as the single crystal diffraction diagrams demonstrate (Fig. 3c). It should be mentioned that after a multi-stage process of microscopy characterization, slicing, RTP treatment, ozone calcination and final conventional calcination, the silicalite-1 monocrystalline slice was analyze by single crystal XRD after each process. This fact indicates the controlled manipulation of the sample. Table 1 shows the high monocrystallinity of the silicalite-1 slice after RTP treatment combined with ozone calcination. A percentage of 89.7% of all the reflections measured matched the calculated orthorhombic unit cell. The remaining reflections were attributed to two other different unit cell components, slightly rotated (0.002 and 0.007°) with respect to the majority component and with respect to each other (0.22°). These components have most of their reflections in common, being almost superimposed.
Conclusions
Calcination using ozone after rapid thermal treatment generated a minor loss of monocrystallinity (about 10%) of giant silicalite-1 samples. However, it provoked a mild distortion in the framework during the thermal treatment to remove the OSDA. The results obtained here are in agreement with the previous research carried out by Choi et al.13 on the improvement of the separation performance of silicalite-1 polycrystalline membranes upon RTP activation.Acknowledgements
This work was supported by the Spanish MINECO (MAT2010-15870), the Aragón Government, and the ESF and FPU Program fellowships (M. N.). We acknowledge the use of Laboratorio de Microscopías Avanzadas facilities at the Instituto de Nanociencia de Aragón (LMA-INA) where the electron microscopy characterization was done. Authors acknowledge the LMA-INA for offering access to their instruments and expertise. The authors would like to acknowledge the use of the Servicio General de Apoyo a la Investigación-SAI (Universidad de Zaragoza).Notes and references
- G. T. Kokotailo, S. L. Lawton, D. H. Olson and W. M. Meier, Nature, 1978, 272, 437 CrossRef CAS.
- J. K. Reddy, K. Motokura, T.-R. Koyama, A. Miyaji and T. Baba, J. Catal., 2012, 289, 53 CrossRef CAS PubMed.
- Z. Lai, G. Bonilla, I. Diaz, J. G. Nery, K. Sujaoti, M. A. Amat, E. Kokkoli, O. Terasaki, R. W. Thompson and M. Tsapatsis, Science, 2003, 300, 456 CAS.
- G. Bonilla, I. Diaz, M. Tsapatsis, H. K. Jeong, Y. Lee and D. G. Vlachos, Chem. Mater., 2004, 16, 5697 CrossRef CAS.
- T. Bein, Chem. Mater., 1996, 8, 1636 CrossRef CAS.
- J. Gascon, F. Kapteijn, B. Zornoza, V. Sebastian, C. Casado and J. Coronas, Chem. Mater., 2012, 24, 2829 CrossRef CAS.
- O. de la Iglesia, P. Hrabanek, A. Zikanova and M. Kocirik, Mater. Lett., 2012, 70, 138 CrossRef CAS PubMed.
- S. B. Tantekin-Ersolmaz, C. Atalay-Orala, M. Tather, A. Erdem-Senatalar, B. Schoeman and J. Sterte, J. Membr. Sci., 2000, 175, 285 CrossRef CAS.
- M. Vilaseca, J. Coronas, A. Cirera, A. Cornet, J. R. Morante and J. Santamaria, Sens. Actuators, B, 2007, 124, 99 CrossRef CAS PubMed.
- C. M. Lew, R. Cai and Y. S. Yan, Acc. Chem. Res., 2010, 43, 210 CrossRef CAS PubMed; M. P. Pina, R. Mallada, M. Arruebo, M. Urbiztondo, N. Navascues, O. de la Iglesia and J. Santamaria, Microporous Mesoporous Mater., 2011, 144, 19 CrossRef PubMed.
- H. Lee, S. I. Zones and M. E. Davis, Nature, 2003, 425, 385 CrossRef CAS PubMed.
- M. Navarro, E. Mateo, B. Diosdado and J. Coronas, CrystEngComm, 2012, 14, 6016 RSC.
- J. Choi, H.-K. Jeong, M. A. Snyder, J. A. Stoeger, R. I. Masel and M. Tsapatsis, Science, 2009, 325, 590 CrossRef CAS PubMed.
- E. Mateo, A. Paniagua, C. Güell, J. Coronas and J. Santamaría, Mater. Res. Bull., 2009, 44, 1280 CrossRef CAS PubMed.
- Y. Li and J. Yu, Chem. Rev., 2014, 114, 7268 CrossRef CAS PubMed.
- S. Shimizu and H. Hamada, Microporous Mesoporous Mater., 2001, 48, 39 CrossRef CAS.
- H. Van Koningsveld, J. C. Jansen and H. Van Bekkum, Zeolites, 1990, 10, 235 CrossRef CAS.
- W. C. Yoo, J. A. Stoeger, P.-S. Lee, M. Tsapatsis and A. Stein, Angew. Chem., Int. Ed., 2010, 49, 8699 CrossRef CAS PubMed.
- J. Kuhn, M. Motegh, J. Gross and F. Kapteijn, Microporous Mesoporous Mater., 2009, 120, 35 CrossRef CAS PubMed; I. Pellejero, M. A. Urbiztondo, M. P. Pina and J. Santamaría, J. Membr. Sci., 2014, 460, 34 CrossRef PubMed.
- O. Pachtova, M. Kocirik, A. Zikanova, B. Bernauer, S. Miachon and J. A. Dalmon, Microporous Mesoporous Mater., 2002, 55, 285 CrossRef CAS.
| This journal is © The Royal Society of Chemistry 2015 |