
Low frequency ultrasound assisted sequential and co-precipitation syntheses of nanoporous RE (Gd and Sm) doped cerium oxide
Abstract
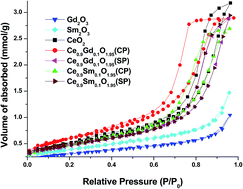
Cerium oxide (CeO2), Ce0.9Gd0.1O1.95 and Ce0.9Sm0.1O1.95 nanostructures were synthesized using 42 kHz ultrasound assisted sequential and co-precipitation techniques. The nanoporous nature of the powders was revealed from the BET analysis which showed that the observed pore size distributions and the H2 hysteresis loops of the respective nanostructures confirmed the existence of nanopores in various sizes and shapes. The nanoporous nature of the synthesized powders were further confirmed by the high resolution transmission electron microscopy (HRTEM) and high angle annular dark field (HAADF) scanning transmission electron microscopy (STEM) analyses. The perceived shift in the characteristic F2g Raman active band centered at 464 cm−1 indicated the defects were increased during the sequential co-precipitation synthesis of Ce0.9Gd0.1O1.95 and Ce0.9Sm0.1O1.95 nanostructures. Moreover, the intensity of the F2g peaks can be ordered as CeO2 < Ce0.9Sm0.1O1.95 (CP)1 < Ce0.9Gd0.1O1.95 (SP)2 < Ce0.9Sm0.1O1.95 (SP)2 < Ce0.9Gd0.1O1.95 (CP)1 which indicates the increase in the concentration of the defects like oxygen vacancies enhances the efficiency of the solid oxide fuel cells (SOFCs) as electrolyte materials. The diffuse reflectance UV-vis solid state spectroscopic analysis demonstrated the narrowing optical band gap of CeO2 during the sequential and co-precipitation of the Ce0.9Gd0.1O1.95 and Ce0.9Sm0.1O1.95 nanostructures.
 Please wait while we load your content...
Please wait while we load your content...

