
XANES, EXAFS and photocatalytic investigations on copper oxide nanoparticles and nanocomposites
Abstract
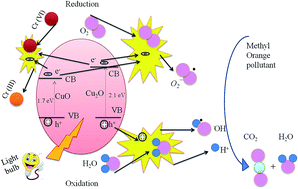
CuO nanoparticles (NPs) and Cu2O/CuO and CuO/TiO2 nanocomposites (NCs) have been synthesized by using a modified co-precipitation method with three different schemes of synthesis. The crystal structures and morphologies of the samples have been investigated using synchrotron X-ray diffraction and transmission electron microscopy, respectively. The detailed local electronic structures of the NPs and NCs have been determined using X-ray absorption near edge structure (XANES) and extended X-ray absorption fine structure (EXAFS) spectroscopy. O K-, Cu K- and Cu L-edge XANES spectra revealed a dominant +2 valence state of Cu in the case of the CuO NPs and CuO/TiO2 NCs, although Cu+1 was dominant in the Cu2O/CuO NCs. A comparison of the local atomic structure around the Cu sites revealed shorter Cu–O bond distances in the as-synthesized samples with respect to the bulk CuO or Cu2O. The Ti K-edge EXAFS fittings for the CuO/TiO2 NCs revealed that the local anatase TiO2 phase was formed, with a Ti–O bond distance of 1.98 Å. We further demonstrated that the CuO NPs, and Cu2O/CuO and CuO/TiO2 NCs can serve as effective photocatalysts towards the degradation of two novel water pollutants, (i) methyl orange (MO) and (ii) potassium dichromate (PD), under visible light irradiation. It was found that the Cu2O/CuO NCs exhibit a higher photocatalytic activity towards the degradation of MO and PD than the CuO NPs or CuO/TiO2 NCs. The mechanism of the photodegradation of MO and PD is also discussed in terms of possible chemical reactions, along with the electronic structure and surface properties of the samples.
 Please wait while we load your content...
Please wait while we load your content...

