
A series of temperature-dependent CdII-complexes containing an important family of N-rich heterocycles from in situ conversion of pyridine-type Schiff base†
Abstract
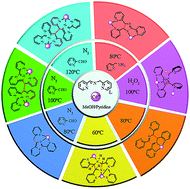
An efficient method for the synthesis of a wide variety of N-rich heterocycles has been systematically explored. The synthetic protocol involves a solvothermal in situ metal–ligand reaction of pyridine-type Schiff base N-(2-pyridylmethyl)-pyridine-2-carbaldimine (L) with Cd2+ ions, resulting in the efficient formation of nine temperature-dependent CdII-complexes 1–9 supported by six types of N-rich heterocycles L1–6. To the best of our knowledge, both the synthetic strategy with solvothermal in situ CdII-mediated Schiff base-conversion and N-rich heterocycle rings L1–2 as well as cis-L6 are reported for the first time. Meanwhile, plausible in situ formation mechanisms of L1–6 are also proposed.
 Please wait while we load your content...
Please wait while we load your content...

