
Self-assembled superparamagnetic nanocomposite-labelled cells for noninvasive, controlled, targeted delivery and therapy†
Ansar Ereath Beeranac,
Francis Boniface Fernandezbc,
Annie Johnbc and
Harikrishna Varma PR*ac
aBioceramics Laboratory, Thiruvananthapuram-695 019, Kerala, India. E-mail: varma@sctimst.ac.in
bTransmission Electron Microscopy Laboratory, Thiruvananthapuram-695 019, Kerala, India
cSree Chitra Tirunal Institute for Medical Sciences and Technology, Thiruvananthapuram-695 019, Kerala, India
First published on 31st March 2015
Abstract
Efficient delivery of cells to targeted sites at optimal concentrations within rational limits of damage to normal tissue is a major challenge for cell delivery. With the help of magnetic nanoparticles binding to the surface of cells, it is possible to manipulate and control cell mobility using an external magnetic field. Here, we demonstrate physical entrapment of magnetic nanocomposites onto cell surfaces and their manipulation by an external magnetic field. Uniformly embedded nano iron oxide particles in a hydroxyapatite crystallite (HAIO) were synthesized via co-precipitation method. Physiochemical and biological evaluation of the above nanocomposite system showed that the HAIO containing 50 wt% iron oxide (HAIO50) possessed excellent magnetic properties and good cytocompatibility. Prussian blue staining and flow cytometric evaluation of cell–material interactions indicated uniform uptake and a dose-dependent interaction. HAIO50 is found to be a novel matrix for use as an effective and cytocompatible avenue for cell separation, evidenced via Coulter analysis as well as fluorescent imaging of live cells. Post-magnetic separation analysis of cell viability via confocal laser scanning microscopy (CLSM) showed the normal structure and proliferation of separated cells. HAIO50 may be used as an efficient matrix for magnetic non-invasive manipulation and for further cell delivery applications.
1 Introduction
Superparamagnetic iron oxide nanoparticles (SPION), due to their superior inherent magnetic properties, have been recognized as a potential candidate for several biomedical applications, such as separation of biomolecular components,1,2 MRI contrast agents,3 hyperthermia4 and controlled drug delivery.5,6 The literature testifies that magnetite and maghemite are the preferred phases of SPION that are more effectively explored for such diverse biological applications.7,8 Magnetic nanoparticle-based cell therapy is one promising area where cells are effectively labelled, separated and concentrated from a biological suspension containing multiple components and delivered to the specific site with the aid of an external magnetic field.9Cell therapy provides more promising solution for several disease and injuries compared to most conventional medicines and therapies, particularly because cells can perform better physiologic as well as metabolic duties than any of the mechanical devices, recombination proteins or chemical compounds.8,10 However, there are a lot of hurdles to systemic administration of bare cells, causing significant difficulties for effective retention of the therapeutic cells at the target site. In order to achieve greater efficiency and optimum performance, a higher cell dose or higher engraftment of cells is inevitable.8,11 Nevertheless, higher cell doses induce larger systemic circulation, which in turn raises safety concerns. Since the fundamental requirement associated with the success of cell therapy is the ability of cells to migrate and engraft,12,13 the inability to achieve the desired level of cell homing and engraftment is a basic challenge for cell-based therapy.
Cells tagged with SPION can migrate easily and enhance accumulation by magnetic actuation.14 Recent literature puts forth several interesting research attempts at magnetic nanoparticle-conjugated stem cell delivery towards tissue repair as well as hyperthermia applications.15–17 Andreas et al. reported citrate-modified SPION-labelled stem cell delivery and its MRI trafficking.18 Kyrtatos et al. reported that ferridex-labelled endothelial progenitor cells efficiently targeted an arterial injury with the help of an external magnetic field.19 Basically, labelling techniques utilize either of the following two approaches: (a) immobilizing magnetic nanoparticles onto the cell surface20 or (b) internalization of biofunctional magnetic nanoparticles, for example via endocytosis.21 In receptor-mediated endocytosis, more particles will have the opportunity to accumulate inside the cells are causes to cell stress.22 Therefore, surface-charge enhanced nanoparticle cell labelling may be considered as a suitable option.
The prerequisites for magnetic nanoparticles to be used for such applications are as follows: should be stable enough to retain its physical integrity, retain its chemical stability, and remain in the suspension state. More significantly, it should not induce any unfavoured reactions in the biological milieu; nevertheless, it should facilitate faster and efficient binding to the required biomolecular component. In addition, feasibility of large-scale production without compromising its fundamental superparamagnetic nature is essential for clinically significant magnetic nanoparticles.
The aggregation of bare SPION is attributed to the van der Waals forces and the magnetic dipolar interactions associated with materials showing ferromagnetic properties.23 Surface functionalization is very essential and is a generally used method to impart long term stability as well as biofunctionality to the nanoparticles.24–26 Among the diverse surface modification techniques, use of an inorganic matrix is recognized as an effective method for imparting uniform particle size to magnetic nanocrystals27,28 by formulating a homogeneous dispersion of the precursors. Recently, the authors developed a superparamagnetic nanocomposite consisting of very uniform size of SPIONs homogenously embedded in hydroxyapatite nanoparticles.29 Also, the biofunctional properties of the iron oxide-incorporated HA have been systematically evaluated, such as cellular interaction, gene delivery, bone regeneration and hyperthermia therapy.30–32 Hydroxyapatite (Ca10(PO4)6(OH)2) has been well recognized as one of the most biocompatible bioceramic materials and has been used for several clinical applications.33,34 Various weight percentage magnetic and non-magnetic phases of compositions of the nanocomposite were synthesized through the co-precipitation route. The various physicochemical characterizations were investigated in order to understand the phase purity, nature of bonding, magnetic property and percentage compositions of the nanocomposite. The present study highlights the unique behaviour of the above nano magnetic bioceramic particles towards some specific cells. Considering magnetic property and biocompatibility of iron oxide (IO) and hydroxyapatite (HA) phases respectively, a composite containing 50–50 wt% of IO and HA [HAIO50] was subjected to in vitro cell–material interaction studies. The various concentrations of HAIO50 were used for cell separation from the biological suspension. HeLa cells were selected for a model cell separation experiment. The role of concentration as well as time in enabling cell labelling was quantitatively evaluated using HAIO50 nanoparticles. The HAIO50-labelled cells were effectively separated and cultured. The design is depicted schematically in Fig. 1. Our results demonstrate an efficient non-invasive mode of cell therapy with SPION-embedded HA with the aid of an external magnetic field.
 | ||
| Fig. 1 Schematic illustration of the magnetic nanocomposite HAIO50-based cell suspension labelling, separation and culturing under in vitro condition. | ||
2 Materials and method
Samples of FeCl2·4H2O (Merck, Darmstadt, Germany), FeCl3 (Merck), Ca(NO3)2·4H2O (Rankem, New Delhi, India), (NH4)H2PO4 (Rankem), and 25% aqueous NH4OH (SD Fine Chemicals, Mumbai, India) and 35% HCl (SD Fine Chemicals) were used as obtained. 3-(4,5-Dimethyl thiazol-2-yl)-2,5-diphenyltetrazolium bromide (Sigma-Aldrich, USA), streptomycin (Invitrogen, USA) and fetal bovine serum (Invitrogen, USA) were used for the MTT assay. All chemicals used for the experiments other than those mentioned in the materials section were obtained from Sigma-Aldrich, USA.2.1 Synthesis of iron oxide embedded hydroxyapatite (HAIO) composite and iron oxide (IO)
The synthesis of HAIOs was carried out by in situ co-precipitation of iron salts and calcium phosphate precursors in alkaline medium at elevated temperature. Briefly, the iron salts solution was freshly prepared in an acidic medium of HCl using FeCl2·4H2O and FeCl3 in a 1![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) :2 ratio. The Ca(NO3)2·4H2O and (NH4)H2PO4 solutions were mixed in proportions to achieve a Ca/P ratio of 1.67. The Ca(NO3)2·4H2O solution was mixed with the iron salt solution with constant stirring. The pH of the above solution was then slowly increased to 11 by adding 25% ammonia solution together with (NH4)H2PO4 over a period of 1 h. The addition and mixing of the reagents were carried out under a N2 atmosphere. The suspension was aged for 24 h at room temperature, after which the precipitate was washed several times with distilled water and centrifuged to achieve neutral pH conditions. The HAIO samples at weight percentage ratios of 10–90 were synthesised using the same method. In ascending order of weight percentage of iron oxide in the composite, the samples are described as HAIO10, HAIO30, HAIO50, HAIO70, and HAIO90. Bare IO was also prepared using a 1:2 ratio of ferrous:ferric chlorides at the same reaction conditions for size, phase purity, and chemical structure comparison.
:2 ratio. The Ca(NO3)2·4H2O and (NH4)H2PO4 solutions were mixed in proportions to achieve a Ca/P ratio of 1.67. The Ca(NO3)2·4H2O solution was mixed with the iron salt solution with constant stirring. The pH of the above solution was then slowly increased to 11 by adding 25% ammonia solution together with (NH4)H2PO4 over a period of 1 h. The addition and mixing of the reagents were carried out under a N2 atmosphere. The suspension was aged for 24 h at room temperature, after which the precipitate was washed several times with distilled water and centrifuged to achieve neutral pH conditions. The HAIO samples at weight percentage ratios of 10–90 were synthesised using the same method. In ascending order of weight percentage of iron oxide in the composite, the samples are described as HAIO10, HAIO30, HAIO50, HAIO70, and HAIO90. Bare IO was also prepared using a 1:2 ratio of ferrous:ferric chlorides at the same reaction conditions for size, phase purity, and chemical structure comparison.
2.2 Physicochemical characterization of HAIOs and IO nanoparticles composite
High-resolution TEM (HRTEM) analysis was performed to evaluate the morphology and crystal size, and for composition analysis. The TEM images and energy dispersive X-ray spectra (EDS) were collected on a JEOL JEM-2010F microscope operated at 300 kV. TEM samples were prepared by dropping highly dispersed HAIO magnetic nanocomposite samples (very low concentration) on to a copper grid then successively drying at room temperature for 24 h. The micro-scale morphology and the composition analysis of the HAIO nanocomposite were investigated with the aid of ESEM (ESEM; Quanta 200, The Netherlands). Samples were prepared by dispersing in distilled water with ultrasonication for 2 minutes. A single drop of the above suspension was put on an aluminium stub and dried at room temperature, followed by coating with gold. The composition of the magnetic nanocomposites was evaluated using energy dispersive X-ray spectroscopy. The phase purity of crystals of HA, IO and HAIOs nanocomposites was analyzed using an X-ray diffractometer (Bruker, D8 Advance, Karlsruhe, Germany) using CuKα1 radiation operating at 40 kV and 30 mA current strength. The crystal structure was determined by analyzing the position and intensities of the diffraction peaks typically observed in the range of diffraction angle 2θ = 20–70° and at a scan rate of 4° min−1 with a step of 0.1°. The hydrodynamic size and surface charge of the nanocomposite particles were analyzed using a Dynamic Light Scattering (DLS) Particle Size Analyzer (Malvern Instruments Ltd, Worcestershire, UK) by dispersing the sample in distilled water using ultrasonic probe sonication. The Fourier transform infrared (FTIR) spectra of the samples were collected using a Thermo-Nicolet 5700 spectrometer. As the ceramic powder was found to be opaque to IR, the diffuse reflectance (DRIFT) technique was used for measurement. Samples were dried and the powder thus obtained was thoroughly mixed with IR grade KBr powder and the reflectance spectrum recorded in the range of 400 to 4000 cm−1 at a resolution of 4 cm−1. KBr powder alone was used for background spectra. Freeze-dried powder samples were used for the magnetic property analysis. Vibrating sample magnetometry (VSM) was used to measure the magnetic properties of the IO and HAIOs using a PAR EG&G Model 4500 magnetometer with an external field varying from −15 to 15 kOe at room temperature. The magnetization of each sample was obtained as a function of the applied field.2.3 In vitro biocompatibility
HeLa (human cervical carcinoma) cells were cultured in Dulbecco's Modified Eagle Medium-High Glucose (DMEM-HG) with 10% fetal bovine serum (FBS), 50 units per ml of penicillin and 50 μg ml−1 of streptomycin. All reagents were sourced from Invitrogen, India and cell culture lab ware from NUNC, Denmark. Cells were seeded and maintained at 37 °C and 5% CO2 atmosphere and experiments were performed at 80% confluence.
The calculated percentages of hemolysis for all the samples were compared with ASTM standard,36 which defines samples as highly hemocompatible (<5% hemolysis), hemocompatible (within 10% hemolysis) and nonhemocompatible (>20% hemolysis).
2.4 Cell separation
HeLa cells (1.4 × 103 cells per μl) were added to seven tubes (A–G) and incubated for 15 minutes with varying concentrations of HAIO50 (C1 to C6) in PBS. Cells were pre-stained with Acridine orange and magnetic separation was carried out with an external magnet (0.3 T) for 1 minute on all tubes. The supernatant and the pellet were collected into separate tubes. Cell numbers were evaluated using Coulter counting (Sysmex K-4500). The supernatant and pellet were resuspended in 500 μl of PBS and placed on a UV transilluminator (Bangalore, Genei).:20 volume dilution). Excess stain was removed by washing with diluted buffer solution, and the sample was then dried and imaged under an inverted phase contrast microscope (Leitz DMIL, Leica, Germany). Environmental scanning electron microscopy (FEI QUANTA 200) was carried out on fixed pellets dehydrated in an ascending alcohol series and placed on a glass coverslip.2.5 Cell culture of magnetic separated cells
Cell pellets collected by magnetic separation were transferred into culture wells under aseptic conditions and provided with growth medium (DMEM-HG) and then cultured for 24 hours. Cell morphology was assessed using a Leica DMIL inverted microscope.3 Results and discussion
The magnetic composite was synthesized using a common wet-precipitation technique that favours the formation of nanoparticles. In principle, iron oxide (IO) nanoparticles are primarily formed and their charged surfaces subsequently initiate the nucleation of amorphous calcium phosphate. This initial calcium phosphate precursor layer is the key step that facilitates the embedding of iron oxide particles, which later on transform to the apatite matrix. In addition, the initial adsorption of calcium phosphate prevents self-agglomeration of IO particles and hence uniformly distributed IO nanoparticles embedded in the calcium phosphate matrix could be achieved. The same experimental conditions were applied for the synthesis of different compositions of magnetic nanocomposites. It should be noted that the weight percentage ratio plays a critical role in determining the fundamental magnetic behaviour and also significantly influences the crystal shape and growth pattern of the particles. Transmission electron and scanning electron microscopic evaluation impart interesting information on the morphological features of the various compositions of magnetic nanocomposite. Fig. 2 and 3 are the respective TEM and SEM micrographs, depicting the acicular or needle shaped crystals of hydroxyapatite containing IO nanoparticles within it. | ||
| Fig. 2 Transmission electron micrographs of various weight percentages of IO embedded HA samples: (a) HAIO10, (b) HAIO20, (c) HAIO30, (d) HAIO40, (e) HAIO50 and (f) higher magnification of (e). | ||
 | ||
| Fig. 3 Scanning electron micrographs of various weight percentages of iron oxide (IO) embedded HA samples (a) HAIO10, (b) HAIO20, (c) HAIO30, (d) HAIO40 and (e) HAIO50. | ||
It was observed that with increased concentration of IO particles, the shapes and crystal growth patterns were altered, transforming from needle to spherical. The lower concentrations of IO of 10, 20 and 30 wt% show acicular nature crystals (as seen from Fig. 2a–c and 3a–c), while higher concentrations of 40 and 50 wt% changed from acicular to spherical shaped HAIO composites (Fig. 2d and e and 3d and e). S-1 Fig. 1a–e and S-2 Fig. 2a–e† present the EDS spectra of various compositions of HAIOs taken from the respective TEM and SEM micrographs. The corresponding peak intensities of iron, calcium and phosphorous elements in the EDS spectra shows good agreement with the formation of all weight percentage compositions. Moreover, the peak intensity ratio for the calcium and phosphorous peaks gives evidence of the formation of hydroxyapatite nanocrystals, irrespective of the presence of IO particles.
The phase pure magnetic nanocomposite formation prepared via this novel synthesis strategy was analysed by XRD as well as FTIR spectroscopy. The XRD patterns of HAIO50 compared with HA and IO are shown in Fig. 4a–c. The XRD pattern of HAIO50 revealed no secondary phases other than those of normal hydroxyapatite and magnetite. The major peaks corresponding to HA (002), (211), (112), (300), (310), (222), and (213) and representative peaks of iron oxide (220), (311), (400), (422), (511), and (440) could be easily viewed from the spectra. The comparison with ICDD card number 01-071-6336 for iron oxide and 00-009-0432 for HA further confirms the presence of the cubic spinal phase of nano iron oxide (Fe3O4) and hexagonal HA crystal structures. The XRD patterns for various weight percentage compositions of HAIOs are depicted in S-3 Fig. 3a–d.† When the percentage of iron oxide increased, the peaks in the spectra broadened and decreased in intensity. This phenomenon was probably due to the small crystallite size and poor crystallinity of synthesized HAIOs.
 | ||
| Fig. 4 X-ray diffraction (XRD) pattern of (a) HA, (b) IO and (c) HAIO50; [HA PDF = 00-009-0432, IO PDF = 01-071-6336]. | ||
The vibrational spectroscopic evaluation of the samples was carried out with FTIR and the results are presented in S-4 Fig. 4a–c.† The FTIR spectrum of the HAIO50 composites has a characteristic peak at 572 cm−1 corresponding to stretching frequency of the Fe–O bond of the Fe3O4 crystals.14 Moreover, the vibration of hydroxyapatite, such as the ν1(P–O) vibration of phosphate, is observed as a peak at ∼962 cm−1. A peak at ∼471 cm−1 is identified as the ν2(O–P–O) vibration of the phosphate group. The peaks observed at ∼1090 and ∼1040 cm−1 have been identified as v3(P–O anti-symmetric) vibrations. The v4 vibrations have been observed at ∼604 and 567 cm−1. These characteristic peaks show the formation of the pure magnetite phase embedded hydroxyapatite nanocomposite. In addition, from analysis of the FTIR spectra various weight percentage compositions of HAIOs, there were no significant differences between the HA peaks and the IO peaks, which demonstrates that the HAIOs retained their structure similar to pure phase HA and IO (S-5 Fig. 5a–d†).
Magnetic measurements of HAIOs measured at room temperature are given in Fig. 5. The hysteresis displays no remanence or coercivity, which shows that the material compositions are superparamagnetic in nature with the absence of a hysteresis curve. The superparamagnetic nature signifies that the magnetic particles embedded in the system consist of single domain nanocrystals and their magnetic moments are aligned in the direction of the applied magnetic field. The bare iron oxide expresses the highest magnetization value (73 emu g−1), and the magnetization value of its corresponding composites decreases with decreasing content of magnetic crystals in the composite nanoparticles. For a safe cell separation application, the nanocomposite should have good magnetic response and stability, which was observed in HAIO50. The HAIO50 has a magnetization value of 23 emu g−1, which is suitable for cell separation application as it shows an optimal response and accumulates in the presence of external magnet. Moreover, the stability of the HAIO50 was measured using zeta potential measurement and the results are presented in S-6 Fig. 6.† The zeta potential value of the particles is −14 mV, which demonstrates the negative surface charge of HAIO50. The charged surface stabilizes the HAIO50 in the colloidal condition by electrostatic repulsion and it retains the dispersed state for a prolonged time.37–39
 | ||
| Fig. 5 Field-dependent magnetization curves (M − H) at 300 K for magnetic composite with compositions of (a) HAIO10, (b) HAIO30, (c) HAIO50, (d) HAIO70, (e) HAIO90 and (f) IO. | ||
The preliminary cytocompatibility of the HAIOs was evaluated by MTT assay and hemocompatibility test. The MTT assay was used to measure cell viability after incubation with the HAIOs. Cells were incubated with test samples for 24 hours and the viability index was measured on a percentage scale based on formazan production. S-7 Fig. 7† shows that HAIO30, HAIO50, HAIO70 and HAIO90 nanoparticles are associated with very low toxicity when concentrations of 0.75 mg ml−1 and 1.5 mg ml−1 were used over a period of 24 hours of exposure. The cytotoxicity of the HAIOs compared with the control values indicated very little variation with percentage compositions and concentrations. It is noteworthy to mention that all the HAIOs concentrations and compositions showed greater than 70% viability. Further it has been noticed that the cells showed excellent viability even at higher concentration (1.5 mg ml−1) of HAIO50 nanocomposite.
A detailed biological evaluation was performed on lower weight percentage IO compositions of HAIOs, particularly due to increased interest associated with such compositions that have lower iron oxide contents with optimal levels of magnetic activity. Haemolysis study was performed to assess the blood compatibility of the candidate materials, since the intravenous route is the most commonly explored way of administration in practical scenarios. A basic concern was the interaction of the negative surface charge of the HAIOs composites with cell membranes, which have an inherent negative charge. The resulting damage, if any, is expressed as percentage of haemoglobin release in S-8 Table 1.†
Three different compositions of iron oxide hydroxyapatite [HAIO10, HAIO30 and HAIO50] at three different concentrations (0.1 mg, 0.3 mg and 0.5 mg) were subjected to hemolytic analysis. The results show that all the HAIOs samples at all concentrations did not cause hemolysis as haemoglobin release levels were found to be well within the acceptable limits of hemocompatibility and cell viability for a biomaterial.36 From these preliminary physicochemical and biological evaluations of HAIOs, the weight percentage ratio of 50:50 (HAIO50) with lower content of IO showed better magnetic property, non-toxicity and blood compatibility, and hence it was selected for further biological evaluations.
Iron staining was carried out to evaluate the presence of magnetic nanoparticles. 120 μg of the HAIO50 nanocomposite was incubated with HeLa cells followed by Prussian blue staining. The presence of iron is demonstrated by a blue colour within cells. The cytoplasmic presence of the blue colour indicates the uptake of particles by HeLa cells. As per S-9 Fig. 8,† positive Prussian blue staining does not affect the morphology of the cells and they retain their native cellular structure in vitro, thus proving to be non-cytotoxic.
Flow cytometric analysis was used to estimate cell–material interactions as a function of time with exposure to different doses of the nanoparticles. Side scatter is generally thought to be related to both the granularity of the cell and the cell mass. The SSC signal is affected by the refractive index of the cytoplasm and the number of organelles present in the cell.40–42 Generally, FSC provides information on the overall size of the cells. Approximately 1 × 106 HeLa cells were treated with HAIO50 at 30, 60, 120, 240, 480 and 960 μg and held for time periods varying from T0 to T15 (minutes). The corresponding cellular interactions were assessed via changes in forward scatter (FSC) and side scatter (SSC). As indicated in Fig. 6 and S-11 Fig. 9,† cell interactions, indicated by an increase in SSC, are related to HAIO50 concentration and incubation time. A control experiment was performed on a non-exposed population of cells, and cells marked as P1 (control) and P2. This helps differentiation of the cells with no internalization of nanoparticles (P1) from those where there was a strong cell–material interaction. At lower concentrations of 30, 60 and 120 μg the granularity changes [P2 = 0.3, 0.6 and 1.2] were comparable to the control population, even after 15 minutes of incubation. However, in cells treated with higher concentrations of HAIO50 of 240, 480 and 960 μg, the FSC was constant but the SSC intensity was higher depending on the incubation time, as presented in S-10 Table 2 and S-12 Table 3.† That is, cells that took up higher doses of nanoparticles showed higher intensities of SSC. FSC is routinely used as a measure of cellular size comparison, irregular cell shape or damage to the cell membrane. It could be presumed from the results that the surface charge enhanced uptake of HAIO50 without adversely affecting the cell cytoskeleton, as confirmed from the FSC intensities. Hence, it can be stated that HAIO50 favours uptake mechanisms by cells and hence could be explored for cell-separation applications based on its inherent magnetic properties (Fig. 7).
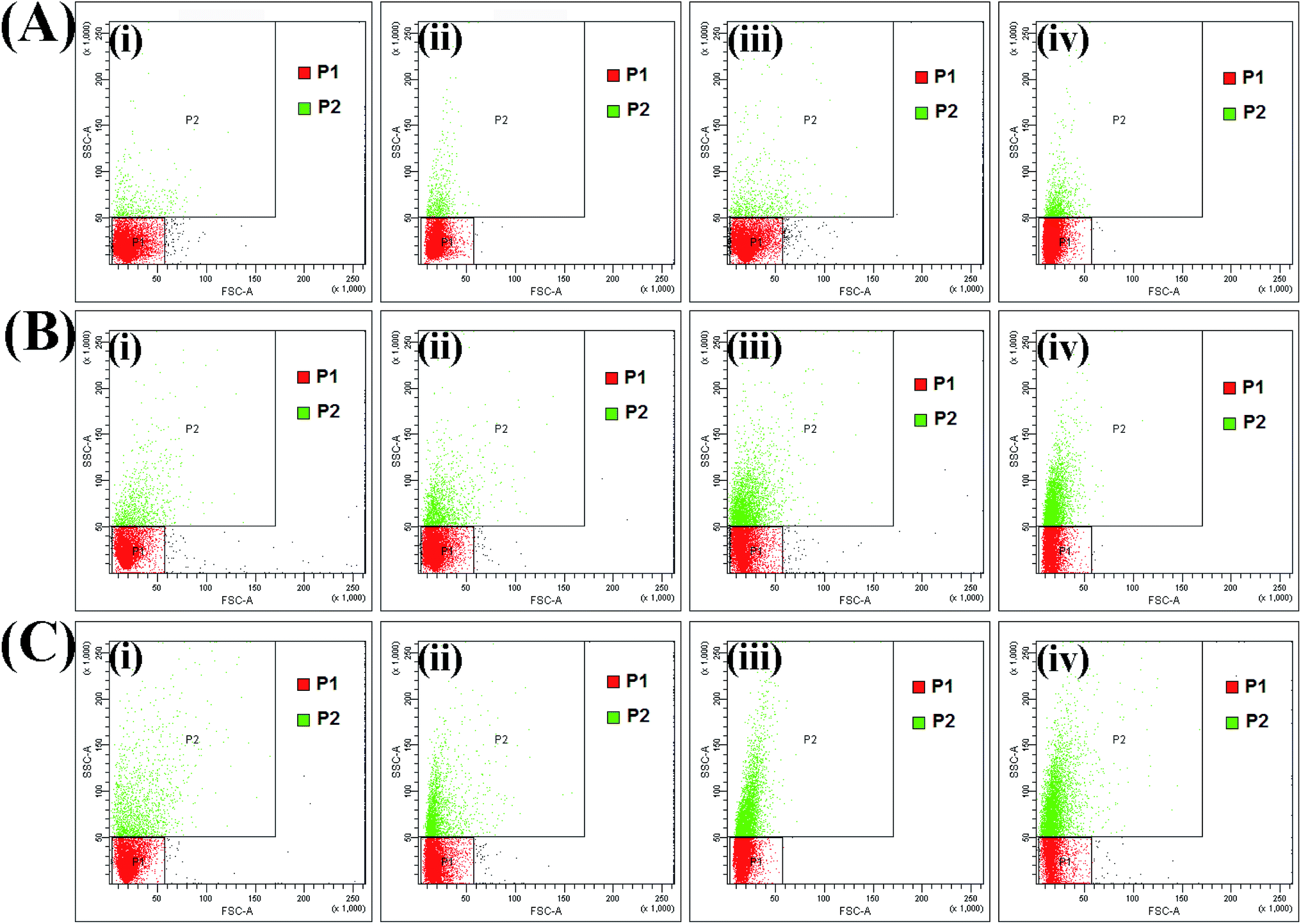 | ||
Fig. 6 FSC vs. SSC plots of flow cytometric measurement of the granularity change in HeLa cells; (A, B and C) show 120 μg, 240 μg and 480 μg of HAIO50, respectively, in contact with 106 cells and (i), (ii), (iii) and (iv) show the analysis at time points 0, 5, 10 and 15 min.  : P1 = cells marked as control indicated by no change in values. : P1 = cells marked as control indicated by no change in values.  : P2 = cells in interaction with HAIO50 indicated by linearly correlated intensity of the SSC channel. Plots indicate an increase in the uptake of HAIO50 from 0 to 15 min and a dose-dependent response at the longer time period with higher dosage. : P2 = cells in interaction with HAIO50 indicated by linearly correlated intensity of the SSC channel. Plots indicate an increase in the uptake of HAIO50 from 0 to 15 min and a dose-dependent response at the longer time period with higher dosage. | ||
 | ||
| Fig. 7 Visible light (A – i, iii, v) and UV (B – ii, iv, vi) illumination of Acridine orange (AO) stained HeLa cells incubated with HAIO50 for 15 min and separated with an external magnet (MT) placed in the vicinity between the 4th and 5th tubes [0.3 T]: M = HAIO50 alone, C + S = cells stained with AO, PBS = buffer, M + C + S = stained cells and HAIO50. (i) and (ii) 0 min, (iii) and (iv) 2 min, and (v) and (vi) 10 min post magnetic exposure. | ||
Flow cytometric analyses results indicate that surface potential enhanced cell binding at the 960 μg level, signifying a possible application in floating cell binding-based magnetic separation. This sets these particles apart from the majority, wherein floating cell separation is achieved using magnetic nanoparticle based receptor–ligand conjugated interaction. To better explore HAIO50 as an efficient probe for floating cell separation from suspension and evaluate its potential as a carrier for cell therapy, low doses of HAIO50 nanoparticles (30, 60, 120, 240, 480, and 960 μg) were incubated with Acridine orange stained 1 × 106 HeLa. The supernatant after magnetic separation as well as the pellet were subjected to cell population analysis using a Coulter Cell Counter. A concentration of 480 μg of HAIO50 was efficiently separated from all cell suspension with a 15 minute incubation period. To further understand the mechanism of interaction with the nanoparticles, separation experiments were carried out at a temperature of 4 °C, which would inhibit energy consuming processes such as direct endocytosis of nanoparticles to the cells.43 The separation efficiency of the 480 μg dose preserved at 4 °C was found to be comparable to that at room temperature (Table 1), indicating an independent interaction mechanism.
| Concentrations of HAIO50 (μg) | Supernatant cell count (HeLa × 103 cells) | |
|---|---|---|
| Temperature (25 °C) | Temperature (4 °C) | |
| 30 | 1.4 | 2 |
| 60 | 0.8 | 1.6 |
| 120 | 0.6 | 1.4 |
| 240 | 0.3 | 0.6 |
| 480 | 0.2 | 0 |
| 960 | 0 | 0 |
As cells are tagged with Acridine orange, to ascertain the fluorescent intensity of supernatant and pellet, the magnetically separated stained HeLa cells were placed under a UV transilluminator. Images in S-13 Fig. 10† show that there was strong green fluorescence in the control tube while fluorescence was absent in the supernatant from tubes B and C, corresponding to 480 μg and 960 μg, respectively. The supernatant from tubes D through G, 240, 120, 60 and 30 μg, respectively, showed increasing levels of fluorescence, which further corroborate the Coulter counter observations of a residual cell population at lower particle concentrations. To confirm cell separation into the pellet compartment, pellets were re-suspended and observed under UV illumination. A dose-dependent decrease in fluorescence with lowering the dose could be visualized in S-13 Fig. 10.†
Cell separation techniques should ideally preserve cell morphology and integrity to help in the continued functionality of the cells post-magnetic separation. In order to evaluate this, magnetically separated cells were observed for their morphology using Giemsa and ESEM techniques.
Giemsa-stained cells were viewed as a dark purple colour under light microscopy, as represented in S-14 Fig. 11,† while the unattached cells on the glass slide were observed as spherical units with a dark violet colour in both the control and the pellet. HAIO50 clumps appeared as a dark yellow colour in both the pellet and the bare sample. The characteristic spherical morphology of the cells was preserved in the control and from cells recovered from the pellet, indicating a cell-friendly separation method. Environmental scanning electron microscopy was further used to evaluate the smears of the control and the pellet recovered cells, as depicted in Fig. 8. Separated cells were similar to the corresponding control cell outlines, while those with adhered nanoparticles were visible on the cell surfaces and confirmed by the corresponding EDS spectrum.
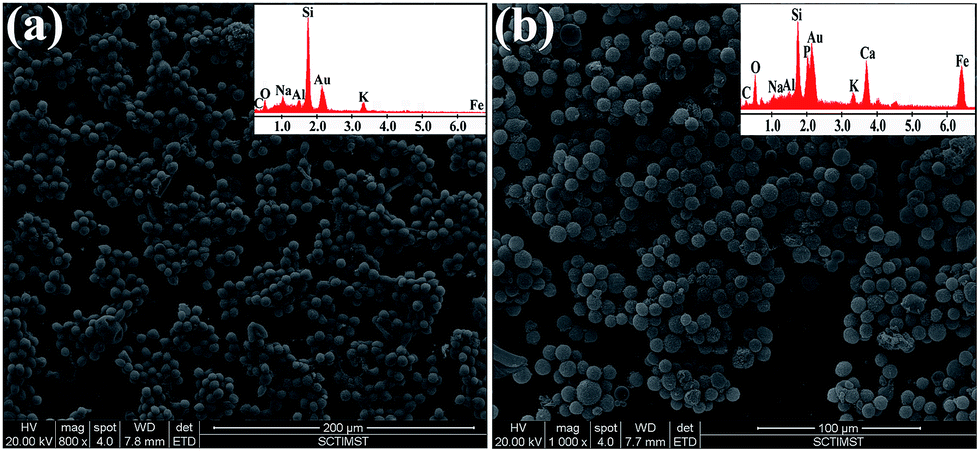 | ||
| Fig. 8 Scanning electron micrographs: (a) cells alone and (b) magnetically separated HAIO50 cell pellet. The corresponding energy dispersive spectra are in the insets. | ||
Through magnetic accumulation-induced cell therapy, an increase in the number of cells accumulating at the injury site is reported,44,45 but it is important to note that a continuous reduction in cell survival and localization at the target site occurs over time in previous studies.11,46 In order to establish the efficacy of magnetic separated cells for cell therapy, and to demonstrate a non-deleterious effect to cellular structure and functions, cells post-separation were maintained in culture. Cells were separated using a 60 μg HAIO50 dose and maintained under standard cell culture conditions for 24 hours post-separation. Actin staining and visualization using confocal laser scanning microscopy studies confirmed the normal cytoplasmic skeletal organization (Fig. 9).
 | ||
| Fig. 9 Confocal laser scanning micrographs of a magnetically separated HAIO50 cell pellet in culture for 24 h (i) in DIC mode, (ii) DAPI (nuclei) stained cells, (iii) rhodamine phalloidin stained actin, and (iv) merged image of (ii) and (iii). (A) Control cells alone and (B) magnetically separated HAIO50 cell pellet. | ||
Individual actin fibres appeared as organized well-defined and clearly visible and HAIO50 particles are viewed as black intracellular spots. The depth and diameter of the cells were calculated from the 3-D reconstructed image of the cells obtained from z-axis scans. From the actin cytoskeleton distribution evaluation, no structural changes were observed in the magnetically separated cells compared to the control cells, and the fibrous structure with cell–cell contact is observed only in biocompatible conditions. Therefore, magnetic composite-based cell separation and subsequent culture could be employed as an efficient technique for cell transplantation therapy.
4 Conclusion
In summary, we designed and fabricated a novel type of intelligent magnetic nanoparticles for targeted cell delivery. The biphasic magnetic nanocomposite consisting of magnetic Fe3O4 embedded in a matrix of hydroxyapatite crystallites was synthesised by base-induced aqueous chemical co-precipitation technique resulting in narrow size distributions. Synthesized HAIOs particles exhibit reliable superparamagnetic properties. Furthermore, the nanocomposite consisting of 50% iron oxide, HAIO50, exhibited good magnetic hysteresis behaviour and biocompatibility, and showed significant cell surface binding properties. Based on evaluations by Coulter counter and UV transilluminator, the capability to reliably separate the cell assembly from suspension was quantified and illustrated. Moreover, the cells with incorporated HAIO50 could be controlled by a non-invasive magnetic field, concentrated separated and cultured under in vitro condition with no detectable impact on cell growth, proliferation or intracellular structures. Our results highlight the potential for using HAIO50-labelled cells as a new type of nanoprobe for remotely controlled cell therapies with better specificity and enhanced efficacy.Acknowledgements
The authors express sincere thanks to the Director, SCTIMST and Head, BMT wing for the facilities provided. In addition, the authors thank the Department of Science and Technology (DST), Govt. of India, for the financial support under the ‘Synthesis of oxide based magnetic nanoparticles for biocompatibility studies, magnetic hyperthermia and MRI application’ program. The support from Dr Manoj Rama Varma, NIIST Trivardrum in the HR-TEM analysis and Dr P. A. Joy, NCL, Pune in the VSM studies are also sincerely acknowledged.References
- H. W. Child, P. A. Del Pino, J. M. De La Fuente, A. S. Hursthouse, D. Stirling, M. Mullen, G. M. McPhee, C. Nixon, V. Jayawarna and C. C. Berry, ACS Nano, 2011, 5, 7910–7919 CrossRef CAS PubMed.
- I. K. Herrmann, M. Urner, F. M. Koehler, M. Hasler, B. Roth-Z'Graggen, R. N. Grass, U. Ziegler, B. Beck-Schimmer and W. J. Stark, Small, 2010, 6, 1388–1392 CrossRef CAS PubMed.
- H. B. Na, I. C. Song and T. Hyeon, Adv. Mater., 2009, 21, 2133–2148 CrossRef CAS PubMed.
- B. Mehdaoui, A. Meffre, J. Carrey, S. Lachaize, L.-M. Lacroix, M. Gougeon, B. Chaudret and M. Respaud, Adv. Funct. Mater., 2011, 21, 4573–4581 CrossRef CAS PubMed.
- Y. Li, J. Ma, H. Zhu, X. Gao, H. Dong and D. Shi, ACS Appl. Mater. Interfaces, 2013, 5, 7227–7235 CAS.
- D. K. Kim, M. Mikhaylova, F. H. Wang, J. Kehr, B. Bjelke, Y. Zhang, T. Tsakalakos and M. Muhammed, Chem. Mater., 2003, 15, 4343–4351 CrossRef CAS.
- Z. R. Stephen, F. M. Kievit and M. Zhang, Mater. Today, 2011, 14, 330–338 CrossRef CAS.
- Y. Wang, Theranostics, 2013, 3, 544–560 CrossRef PubMed.
- O. Olsvik, T. Popovic, E. Skjerve, K. S. Cudjoe, E. Hornes, J. Ugelstad and M. Uhlen, Clin. Microbiol. Rev., 1994, 7, 43–54 CAS.
- D. J. Mooney and H. Vandenburgh, Cell Stem Cell, 2008, 2, 205–213 CrossRef CAS PubMed.
- S.-H. Li, T. Y. Y. Lai, Z. Sun, M. Han, E. Moriyama, B. Wilson, S. Fazel, R. D. Weisel, T. Yau, J. C. Wu and R.-K. Li, J. Thorac. Cardiovasc. Surg., 2009, 137, 1225–1233 CrossRef PubMed.
- J. P. Singh, JACC: Cardiovascular Interventions, 2009, 2, 803–804 CrossRef PubMed.
- Z. Huang, N. Pei, Y. Wang, X. Xie, A. Sun, L. Shen, S. Zhang, X. Liu, Y. Zou, J. Qian and J. Ge, Biomaterials, 2010, 31, 2130–2140 CrossRef CAS PubMed.
- J. Chen, N. Huang, B. Ma, M. F. Maitz, J. Wang, J. Li, Q. Li, Y. Zhao, K. Xiong and X. Liu, ACS Appl. Mater. Interfaces, 2013, 5, 5976–5985 CAS.
- M. Edmundson, N. T. Thanh and B. Song, Theranostics, 2013, 3, 573–582 CrossRef PubMed.
- S. Kubinová and E. Syková, Nanomedicine, 2010, 5, 99–108 CrossRef PubMed.
- J. W. M. Bulte, S.-C. Zhang, P. van Gelderen, V. Herynek, E. K. Jordan, I. D. Duncan and J. A. Frank, Proc. Natl. Acad. Sci. U. S. A., 1999, 96, 15256–15261 CrossRef CAS.
- K. Andreas, R. Georgieva, M. Ladwig, S. Mueller, M. Notter, M. Sittinger and J. Ringe, Biomaterials, 2012, 33, 4515–4525 CrossRef CAS PubMed.
- P. G. Kyrtatos, P. Lehtolainen, M. Junemann-Ramirez, A. Garcia-Prieto, A. N. Price, J. F. Martin, D. G. Gadian, Q. A. Pankhurst and M. F. Lythgoe, JACC: Cardiovascular Interventions, 2009, 2, 794–802 CrossRef PubMed.
- R. Handgretinger, P. Lang, M. Schumm, G. Taylor, S. Neu, E. Koscielnak, D. Niethammer and T. Klingebiel, Bone Marrow Transplant., 1998, 21, 987–993 CAS.
- U. Schoepf, E. M. Marecos, R. J. Melder, R. K. Jain and R. Weissleder, BioTechniques, 1998, 24, 642 CAS.
- L. Kou, J. Sun, Y. Zhai and Z. He, Asian J. Pharm. Sci., 2013, 8, 1–10 CrossRef CAS PubMed.
- S. Manju, C. P. Sharma and K. Sreenivasan, J. Mater. Chem., 2011, 21, 15708–15717 RSC.
- S. Liang, Y. Wang, J. Yu, C. Zhang, J. Xia and D. Yin, J. Mater. Sci.: Mater. Med., 2007, 18, 2297–2302 CrossRef CAS PubMed.
- A. K. Gupta and M. Gupta, Biomaterials, 2005, 26, 3995–4021 CrossRef CAS PubMed.
- A.-H. Lu, E. L. Salabas and F. Schüth, Angew. Chem., Int. Ed., 2007, 46, 1222–1244 CrossRef CAS PubMed.
- D. Dupont, W. Brullot, M. Bloemen, T. Verbiest and K. Binnemans, ACS Appl. Mater. Interfaces, 2014, 6, 4980–4988 CAS.
- E. M. Moreno, M. Zayat, M. P. Morales, C. J. Serna, A. Roig and D. Levy, Langmuir, 2002, 18, 4972–4978 CrossRef CAS.
- E. B. Ansar, M. Ajeesh, Y. Yokogawa, W. Wunderlich and H. Varma, J. Am. Ceram. Soc., 2012, 95, 2695–2699 CrossRef CAS PubMed.
- H.-C. Wu, T.-W. Wang, M. C. Bohn, F.-H. Lin and M. Spector, Adv. Funct. Mater., 2010, 20, 67–77 CrossRef CAS PubMed.
- N. Tran and T. J. Webster, Acta Biomater., 2011, 7, 1298–1306 CrossRef CAS PubMed.
- C.-H. Hou, S.-M. Hou, Y.-S. Hsueh, J. Lin, H.-C. Wu and F.-H. Lin, Biomaterials, 2009, 30, 3956–3960 CrossRef CAS PubMed.
- H. V. Easwer, A. Rajeev, H. K. Varma, S. Vijayan and R. N. Bhattacharya, Acta Neurochir., 2007, 149, 481–485 CrossRef CAS PubMed , discussion 485–486.
- S. V. Dorozhkin and M. Epple, Angew. Chem., Int. Ed., 2002, 41, 3130–3146 CrossRef CAS.
- T. Mosmann, J. Immunol. Methods, 1983, 65, 55–63 CrossRef CAS.
- S. K. Roy Chowdhury, A. C. Kulkarni, A. Basak and S. K. Roy, Wear, 2007, 262, 1387–1398 CrossRef PubMed.
- M. R. Jahn, H. B. Andreasen, S. Fütterer, T. Nawroth, V. Schünemann, U. Kolb, W. Hofmeister, M. Muñoz, K. Bock, M. Meldal and P. Langguth, Eur. J. Pharm. Biopharm., 2011, 78, 480–491 CrossRef CAS PubMed.
- D.-H. Kim, Y. Guo, Z. Zhang, D. Procissi, J. Nicolai, R. A. Omary and A. C. Larson, Adv. Healthcare Mater., 2013, 3, 714–724 CrossRef PubMed.
- J. J. Thomas, M. R. Rekha and C. P. Sharma, Colloids Surf., B, 2010, 81, 195–205 CrossRef CAS PubMed.
- A. Tzur, J. K. Moore, P. Jorgensen, H. M. Shapiro and M. W. Kirschner, PLoS One, 2011, 6, e16053 CAS.
- H. B. Steen, Cytometry, Part A, 2004, 57, 94–99 CrossRef PubMed.
- R. M. Zucker, E. J. Massaro, K. M. Sanders, L. L. Degn and W. K. Boyes, Cytometry, Part A, 2010, 77, 677–685 CrossRef CAS PubMed.
- V. Sokolova, D. Kozlova, T. Knuschke, J. Buer, A. M. Westendorf and M. Epple, Acta Biomater., 2013, 9, 7527–7535 CrossRef CAS PubMed.
- B. Polyak, I. Fishbein, M. Chorny, I. Alferiev, D. Williams, B. Yellen, G. Friedman and R. J. Levy, Proc. Natl. Acad. Sci. U. S. A., 2008, 105, 698–703 CrossRef CAS PubMed.
- J. Riegler, J. A. Wells, P. G. Kyrtatos, A. N. Price, Q. A. Pankhurst and M. F. Lythgoe, Biomaterials, 2010, 31, 5366–5371 CrossRef CAS PubMed.
- J. V. Terrovitis, R. R. Smith and E. Marbán, Circ. Res., 2010, 106, 479–494 CrossRef CAS PubMed.
Footnote |
| † Electronic supplementary information (ESI) available. See DOI: 10.1039/c4ra16185h |
| This journal is © The Royal Society of Chemistry 2015 |