
Spectroscopic studies of keto–enol tautomeric equilibrium of azo dyes
Abstract
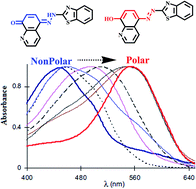
Azo dyes account for 60–70% of all dyes known to date. Understanding the factors that affect the direction of keto–enol tautomerism in azo dyes through spectral measurements is crucial to their potential applications. This review encompasses the most important spectroscopic studies of different azo dyes categorized by their structures within the last few years (2010–2014). It is concluded that the stability of the keto and enol forms largely arises from the ability to establish intra-molecular and inter-molecular hydrogen bonding, respectively. There are many factors that affect the keto or enol form, for example, polar solvent, high temperature, neutral pH and electron withdrawing substituents favor the keto form through intramolecular hydrogen bonding, whereas, nonpolar solvent, low temperature, high pH and electron donating groups favor the enol form through intermolecular hydrogen bonding. Encapsulation inside nanocaged microheterogenous systems creates a rigid environment that stabilizes the enol form mostly, while in the solid state, most of the tautomeric equilibrium lies in favor of the keto form. Understanding of keto–enol tautomerism in azo dyes helps to probe solvation dynamics, to tune the pKa values in chemical sensing, and to explain proton transfer in the excited state.
 Please wait while we load your content...
Please wait while we load your content...

