
Insights into the catalytic mechanism of chlorophenol 4-monooxygenase: a quantum mechanics/molecular mechanics study†
Abstract
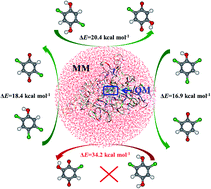
The catalytic mechanism of chlorophenol 4-monooxygenase (TftD) toward substrates 2,4,5-trichlorophenol (2,4,5-TCP), 2,4,6-trichlorophenol (2,4,6-TCP), and 2,5-dichloro-p-hydroquinone (2,5-DiCHQ) was studied by quantum mechanical/molecular mechanical (QM/MM) investigations. The hydroxylation rather than dechlorination was found to be the rate determining step for all three substrates. Substantial energy barrier spreads on the hydroxylation steps have been found. The corresponding Boltzmann-weighted average barriers are 16.3, 17.8, and 19.7 kcal mol−1 for 2,4,5-TCP, 2,4,6-TCP, and 2,5-DiCHQ, in accordance with the experimentally determined reaction rates. A previously unnoticed residue Arg100 was found to be important during the hydroxylation steps. Electrostatic influences of ten active site residues on the hydroxylation step were also investigated. Glu251 and Arg366 were highlighted as candidate residues for future mutation studies. In addition, we have shown that spatial location of 2,6-DiCHQ as well as the hydroxylation barrier are the two possible reasons why 2,6-DiCHQ cannot be further degraded by TftD.
 Please wait while we load your content...
Please wait while we load your content...

