
Gold nanoparticles supported on magnesium ferrite and magnesium oxide for the selective oxidation of benzyl alcohol
Abstract
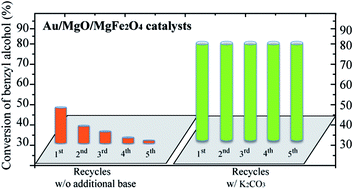
Au nanoparticles (Au NPs) have gained significant attention as catalysts for the selective oxidation of alcohols; however, the catalytic activity is highly dependent on the presence of a base. Alternatives to the use of strong bases, such as NaOH, are still needed. Here, we explored the basicity of magnesium ferrite/oxide supports to study the catalytic behaviour of supported Au NPs for the oxidation of benzyl alcohol. The presence of Mg2+ ions in the ferrite structure improved the catalytic activity of supported Au NPs to ca. 35% conversion in the absence of an additional base. After modifying the support with MgO, the catalytic activity of supported Au NPs was further improved to ca. 50% conversion, but the catalyst deactivated in successive recycling tests. When the catalysts were tested in the presence of a sub-stoichiometric amount of K2CO3, they became more active and remained stable upon recycling with no loss of activity and selectivity for the preferential production of benzoic acid.
 Please wait while we load your content...
Please wait while we load your content...

