
One-step synthesis of three-dimensional porous ionic liquid–carbon nanotube–graphene gel and MnO2–graphene gel as freestanding electrodes for asymmetric supercapacitors†
Yimin Sun*a,
Yubo Chenga,
Kui Hea,
Aijun Zhoua and
Hongwei Duan*b
aSchool of Material Science and Engineering, Wuhan Institute of Technology, Wuhan, Hubei 430073, People's Republic of China. E-mail: ymsun1982@hotmail.com
bSchool of Chemical and Biomedical Engineering, Nanyang Technological University, 70 Nanyang Drive 637457, Singapore. E-mail: hduan@ntu.edu.sg
First published on 6th January 2015
Abstract
We report the design of a new type of high-performance asymmetric supercapacitor using three-dimensional (3D) porous ionic liquid (IL)–carbon nanotube (CNT)–graphene gel and MnO2–graphene gel as freestanding electrodes, both of which are synthesized by a facile and green one-step hydrothermal method. For the synthesis of IL–CNT–graphene gel, the precursor CNT–graphene oxide has firstly been dispersed by IL and then self-assembled into a unique “skeleton/skin” structure with CNTs as the skeleton and graphene nanosheets as the skin. For the synthesis of MnO2–graphene gel, the nucleation and growth of MnO2 nanoparticles as well as the self-assembly of graphene nanosheets occur simultaneously under the hydrothermal treatment, lead to a distinct “plum pudding” structure, where the MnO2 nanoparticles that are decorated on the 3D graphene gel are tightly wrapped by graphene nanosheets. In these two strategies, the introduction of IL, CNT and MnO2 nanoparticles into graphene gel not only acts as the “spacer” to prevent the π–π stacking interactions between graphene nanosheets, but also improves the electrochemical and capacitive properties of the graphene nanohybrid gel. The resultant asymmetric supercapacitor of IL–CNT–graphene gel//MnO2–graphene gel achieves high energy as well as good cycling stability and affordability, and can be reversibly charged/discharged at a maximum cell voltage of 1.8 V in 1.0 M aqueous Na2SO4 electrolyte. The corresponding energy density and power density are 25.6 W h kg−1 and 9.07 kW kg−1 at 1 A g−1, respectively, and the specific capacitance retention remains about 90% after 10![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) 000 cycles.
000 cycles.
Introduction
Supercapacitors (SCs) have attracted considerable attention due to their significant, inherent advantages, such as high energy and power density, good operational safety, and long cycling life as well as low cost and environmental friendliness, in comparison with the current energy storage devices.1 SCs show large specific capacitance based on two mechanisms that occur at or near the electrode/electrolyte interfaces in the capacitors. The first is the electrochemical double layer capacitor (EDLC) mechanism, which stores the charge electrostatically using reversible adsorption of ions of the electrolyte onto active materials on the electrode. The other is the pseudo-capacitor mechanism, which is a Faradic process, using fast and reversible surface or near surface reactions for charge storage. Compared with batteries, SCs, especially EDLCs, are essentially maintenance-free, possess a longer cycle-life, require a very simple charging circuit, experience no memory effect, and are generally much safer. However, they currently suffer from a major limitation of low energy density, which is still much less than that of advance batteries.2 There are tremendous interests in increasing the energy density and making it comparable with that of Li-ion batteries. Technically, enhancements in the energy density of SC can be achieved by increasing either its specific capacitance or its operating voltage.3 Increasing evidences have shown that developing hybrid electrode materials by combining EDLCs and pseudo-capacitance,4–6 using ionic liquids with a wider voltage window as electrolyte,7 utilizing active materials with larger specific surface area, such as microporous and mesoporous materials, nanowire arrays, nanoflakes, nanospheres,8 are the possible solutions.Up-to-date, the most attractive materials for EDLCs are the carbonaceous nanostructures, such as active carbon, carbon fiber, carbon nanotube (CNT) and graphene,9 while the transition metal oxides and conductive polymer are intriguing candidates for pseudocapacitors.10 Among different carbon materials, graphene has attracted most attention owing to their unique two-dimensional structure, remarkable chemical stability, high surface area (calculated theoretical value 2630 m2 g−1), and high electrical conductivity (103 to 104 S m−1).11 Recent research shows that graphene nanosheets can be fabricated or assembled into functional gels with desired three-dimensional (3D) interconnected porous microstructures, such as graphene hydrogel, graphene foam and graphene nanomesh.12 The as-obtained 3D functional graphene materials possess multi-dimensional conductivity, low mass transport resistance, abundant hierarchically porous architectures, large surface area and excellent mechanical/chemical stability, which are highly desirable for SC application.13 However, the graphene sheets usually trend to aggregate due to their strong π–π interaction during the self-assembly process. When used for SCs, the aggregation of graphene nanosheets will hinder the rapid electrolyte diffusion and reduce the surface area of the electrode, and consequently decrease the overall performance of the SC device. To circumvent this challenge, one approach is to blend graphene sheets with other composite materials as “spacers” (e.g., polymers or nanoparticles) to prevent aggregation of graphene sheets by forming composites.14 With the assistance of those active “spacers” materials, graphene sheets can assemble into macroscopic 3D porous structure with high specific surface area and improved mechanical/chemical properties. However, it is still lack of a comprehensive solution to structurally integrate them together rather than simple physical mixing or avoid the sacrifices in electrochemical conductivity and capacitive properties that have limited the success of SCs.
In this work, we adopt two strategies to improve the supercapacitive properties of the graphene-based materials, one is to synthesize a new type of ionic liquid (IL)–CNT–graphene gel, another is to prepare MnO2 nanoparticles decorated graphene gel, both of which are obtained by a facile and green one-step hydrothermal approach, as illustrated in Scheme 1. In the first strategy, the precursor CNT–GO is firstly dispersed by IL to prevent the aggregation of CNT and the closely-contacted packing of graphene sheets. ILs have attracted much attention as promising green solvents because of their unique physicochemical characters such as relatively low viscosity, negligible vapor pressure, effective nonvolatility, thermal stability, increased electrochemical window, and relatively high ionic conductivity.15 ILs are also good solvents for a number of inorganic and organic materials as well as polymers.13 Very recently, poly(ionic liquid) (PIL) has been employed to stabilize graphene platelets via electrostatic interactions, resulting in the formation of PIL-modified graphene materials with outstanding supercapacitive properties.16 In our work, the precursor IL–CNT–GO has self-assembled into a unique “skeleton/skin” structural IL–CNT–graphene gel with CNTs as the skeleton and graphene sheets as the skin. In the second strategy, MnO2 nanoparticles are in situ grown on 3D graphene gel to form MnO2–graphene gel with a distinct “plum pudding” structure under the hydrothermal treatment, by which the nucleation and growth of MnO2 nanoparticles as well as the self-assembly of graphene sheets occur simultaneously, and the MnO2 nanoparticles that decorated on the graphene gel are tightly wrapped by graphene sheets. In these two strategies, the introduction of IL, CNTs and MnO2 nanoparticles not only acts as the “spacer” to prevent the π–π stacking interactions between graphene nanosheets, but also improves the electrochemical and capacitive properties of the resultant graphene nanocomposite. Moreover, the proposed one-step hydrothermal approach does not necessitate any chemical reducing or capping agents, representing a facile and green method to prepare graphene nanocomposite with highly structural integrity and stability.
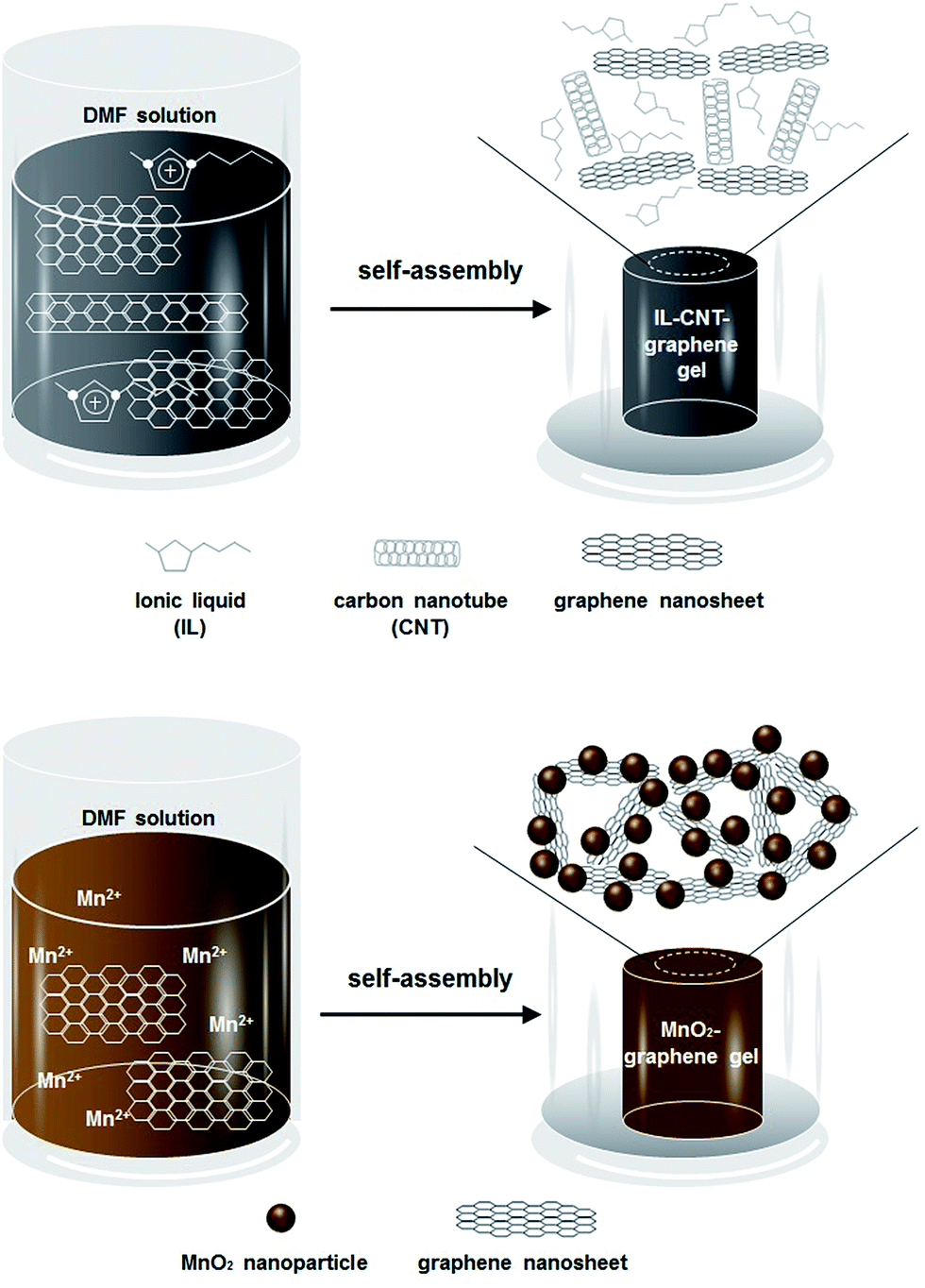 | ||
| Scheme 1 Schematic illustration of the fabrication process of macroscopic IL–CNT–graphene gel and MnO2–graphene gel cylinders. | ||
Owing to the exceptional electrical conductivity and mechanical strength of IL–CNT–graphene gel and MnO2–graphene gel, they can be used as freestanding and binder-free electrodes for SC application. Here an asymmetric SC has been fabricated with IL–CNT–graphene gel and MnO2–graphene gel as the negative electrode and positive electrode, respectively. By utilizing both Faradaic and non-Faradaic processes to store charge, the obtained asymmetric SC of IL–CNT–graphene gel//MnO2–graphene gel combines the advantages of EDLCs and pseudocapacitors, and therefore can achieve high energy and power densities as well as good cycling stability and affordability. The asymmetric SC can be reversibly charged/discharged at a maximum cell voltage of 1.8 V in 1.0 M aqueous Na2SO4 aqueous electrolyte. The corresponding energy density and power density are 25.6 W h kg−1 and 9.07 kW kg−1 at 1 A g−1, respectively, and the specific capacitance retention remains about 90% after 10000 cycles. These encouraging results show great application potential of the proposed asymmetric SC in developing energy storage devices with high energy and power densities for a wide spectrum of commercial viability.
Results and discussion
Physicochemical characterization
GO nanomaterials were synthesized from graphite powder based on Hummer's method, which have abundant polar surface functional groups on their surface. Consequently, the GO nanomaterials are water-dispersible, as shown in the inset of Fig. 1A. The scanning electron microscope (SEM) analysis reveals that the GO nanosheets exhibits typical winkle feature (Fig. 1A). From higher resolution images, it can be observed that the GO nanosheets are corrugated to form a silk veil-like wave structure that consisted of ultrathin stacked nanoflakes (Fig. 1B). The 3D porous reduced GO (rGO) gel was prepared by thermal treatment of initial GO precursor, which can self-assemble into a well-formed black gel cylinder during the gelation process (Fig. 1C inset). It has been recognized that the noncovalent interactions, including π–π stacking interactions between graphene nanosheets, as well as hydrogen bond interactions between the –OH and –COOH groups on graphene nanosheets, are the driven force in the gelation process.13 After freeze-dried in vaccum, the as-obtained rGO gel shows desirable interconnected porous structure (Fig. 1C), the pore sizes within the network are at micrometer scale (Fig. 1D). Nevertheless, the graphene nanosheets are trend to restack during the self-assembly process, which inevitably decrease the surface area of the resultant rGO gel.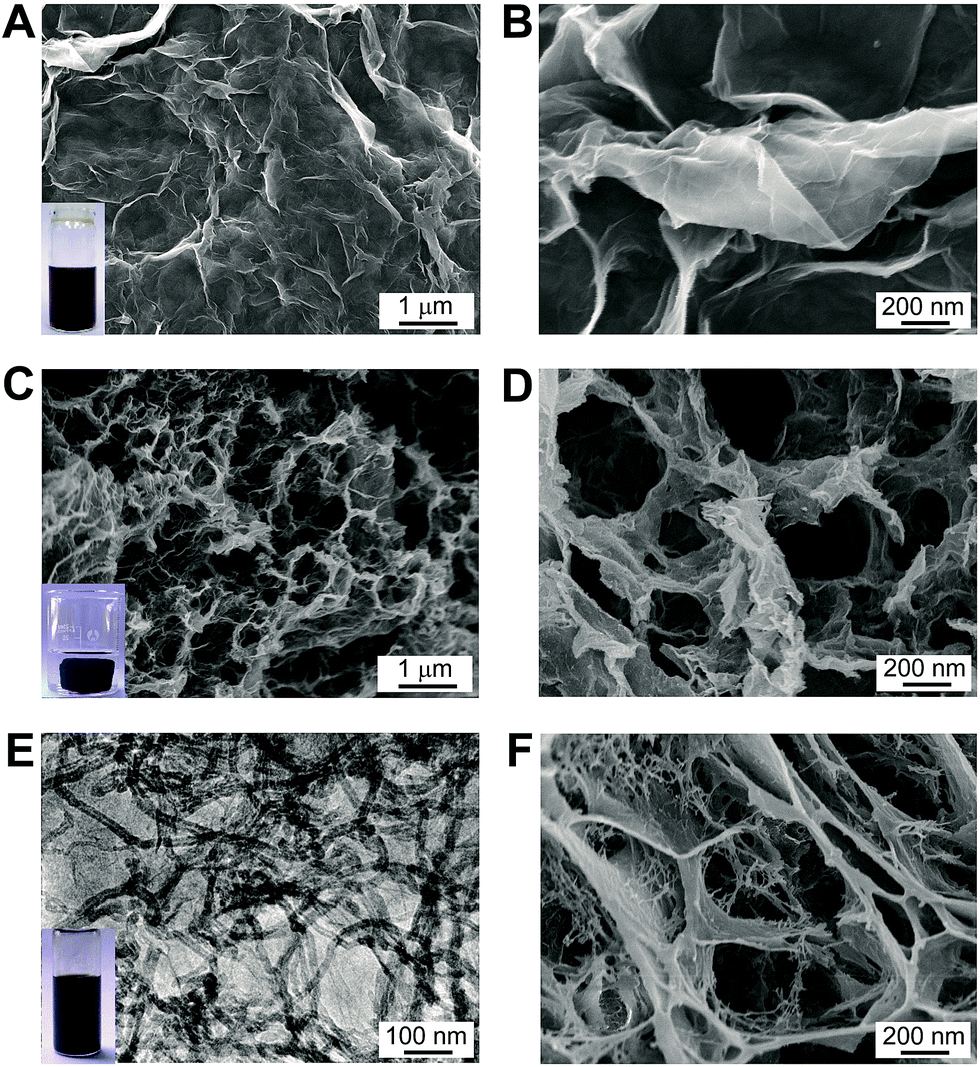 | ||
| Fig. 1 (A) and (B) SEM images of GO nanosheets with different magnifications. Inset of (A) is the photograph of GO suspension; (C) and (D) SEM images of 3D rGO gel with different magnifications. Inset of (C) is the photograph of macroscopic rGO gel cylinder. (E) TEM image of IL–CNT–rGO suspension. Inset of (E) is the photograph of IL–CNT–GO suspension. (F) SEM image of IL–CNT–rGO gel at high magnification. | ||
In order to increase the surface area and improve the electrochemical properties of the 3D graphene-based material, we develop a new type of IL–CNT–rGO gel. The precursor IL–CNT–GO was firstly synthesized by grinding GO powder with CNT and IL (i.e., 1-butyl-3-methylimidazolium tetrafluoroborate, BMIMBF4), upon which the IL, CNT and GO powder can form a uniform black paste. The resultant IL–CNT–GO nanocomposite could be readily redispersed in water to form a homogeneous and stable solution (Fig. 1E inset), in which both CNTs and graphene layer are well-dispersed, as shown in the TEM image (Fig. 1E). This is probably due to the strong interaction between graphene, CNT and IL. The IL with imidazolium ring can interact strongly with the graphitic surface of the graphene and CNT via cation–π interaction, and act as a “spacer” to prevent their disorder stacking and aggregation.17 In addition, the graft of hydrophilic IL (BMIMBF4) to the rGO and CNT plane provides a highly stable dispersibility of graphene and CNT in water.
IL–CNT–rGO gel was prepared by thermal treatment of IL–CNT–GO precursor, which also exhibits 3D porous structure, where the graphene nanosheets are supported by CNT to form a “skeleton/skin” architecture with CNTs as the skeleton and graphene nanosheets as the skin (Fig. 1F). This unique structure, combining with the effect of IL spacers that attach on CNT and graphene nanosheets, effectively increase the surface area of IL–CNT–rGO nanocomposite. The structural feature of IL–CNT–rGO gel was further characterized by nitrogen adsorption and desorption measurements. The adsorption–desorption curve exhibits a distinct reversible hysteresis loop (ESI, Fig. S1A†). Moreover, the corresponding pores size distribution (PSD) of IL–CNT–rGO gel presents a relatively narrow mesopore feature centered around a mesopore entrance size of 3.6 nm (inset in Fig. S1A†), which is advantageous to the transport and diffusion of electrolyte ions during the rapid charge/discharge process.5 The Brunauer–Emmett–Teller (BET) surface area of IL–CNT–rGO gel estimated from Barret–Joyner–Halenda (BJH) analysis of the isotherms is up to 285 m2 g−1. The pore volume and BJH desorption average pore diameter of IL–CNT–rGO gel are 0.966 cm3 g−1 and 13.6 nm, respectively.
MnO2–rGO gel was prepared by one-step self-assembly method, using GO and Mn(CH3COO)2 as the precursors. Under the solvothermal treatment, the nucleation and growth of MnO2 nanoparticles as well as the self-assembly of graphene sheets occur simultaneously, leading to the formation of a unique “plum pudding” structure (Fig. 2A and B), where MnO2 nanoparticles are well distributed in 3D rGO gel and wrapped by graphene nanosheets (Fig. 2C). Although the exact growth mechanism has not been completely understood, it can be deduced that oxygen containing functional groups on graphene can act as anchoring or nucleation sites for the growth of MnO2 nanoparticles. The Mn2+ ions in solution are preferentially adsorbed on them due to the electrostatic force between Mn2+ ions and polar oxygen functional groups.18 The spherical MnO2 are uniform in size with the diameter of 50–100 nm, as shown in the transmission electron microscopic (TEM) image of Fig. 2D. The as-obtained MnO2 nanoparticles are firmly anchored on graphene nanosheets to prevent them from aggregation, which increase the surface area of the resultant MnO2–rGO gel. The BET surface area of MnO2–rGO gel estimated from BJH analysis is 293 m2 g−1. The pore volume and BJH desorption average pore diameter of MnO2–rGO gel are 0.607 cm3 g−1 and 8.29 nm, respectively (Fig. S1B†). And the PSD of MnO2–rGO gel also shows a mesopore size of 3.4 nm with a narrow distribution (inset in Fig. S1B†).
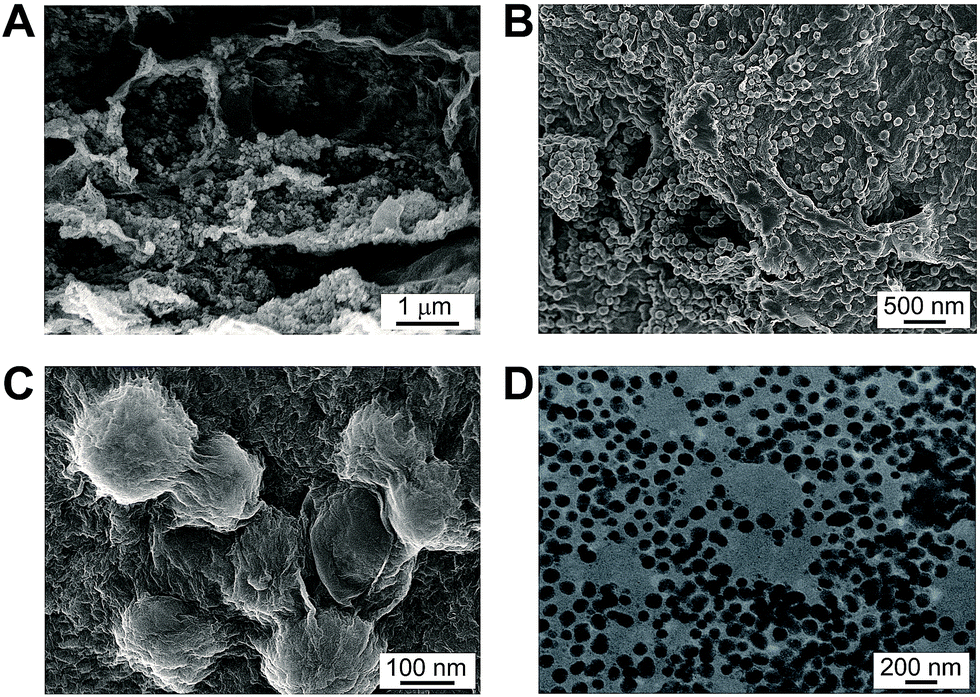 | ||
| Fig. 2 (A–C) SEM images of MnO2–rGO gel with different magnifications. (D) TEM image of MnO2 nanoparticles that detached from MnO2–rGO nanocomposite. | ||
X-ray photoelectron spectroscopy (XPS) has further been employed to intuitively evaluate the reduction level and determine the composition of the as-prepared nanohybrid samples. As shown in Fig. 3A, the curve fits of C1s spectra of GO shows four distinct peaks centered at 285.2, 287.1, 288.5 eV and 289.3 eV, which are corresponding to C![[double bond, length as m-dash]](https://www.rsc.org/images/entities/char_e001.gif) C/C–C in aromatic rings, C–O in epoxy and hydroxyl, CO and O–CO in carboxyl, respectively. While for IL–CNT–rGO nanocomposite, the intensities of all C1s peaks of the carbons binding to oxygen, especially the peak of C–O (epoxy and hydroxyl), decrease dramatically compared to those of GO (Fig. 3B), revealing that most oxygen containing functional groups are removed under the thermal treatment.19 Additionally, a new peak assigned to the C–N bound appears at 285.8 eV, and N1s signal appears at 401.5 eV (Fig. 3C), which are originated from IL (BMIMBF4) that attached on graphene and CNT. Moreover, in the Mn2p core-level signals of MnO2–rGO nanocomposite, Mn2p3/2 and Mn2p1/2 spin–orbit peaks appear at 642.2 and 652.9 eV (Fig. 3D), indicating that element Mn in the prepared samples is present in the chemical state of Mn(IV).20
C/C–C in aromatic rings, C–O in epoxy and hydroxyl, CO and O–CO in carboxyl, respectively. While for IL–CNT–rGO nanocomposite, the intensities of all C1s peaks of the carbons binding to oxygen, especially the peak of C–O (epoxy and hydroxyl), decrease dramatically compared to those of GO (Fig. 3B), revealing that most oxygen containing functional groups are removed under the thermal treatment.19 Additionally, a new peak assigned to the C–N bound appears at 285.8 eV, and N1s signal appears at 401.5 eV (Fig. 3C), which are originated from IL (BMIMBF4) that attached on graphene and CNT. Moreover, in the Mn2p core-level signals of MnO2–rGO nanocomposite, Mn2p3/2 and Mn2p1/2 spin–orbit peaks appear at 642.2 and 652.9 eV (Fig. 3D), indicating that element Mn in the prepared samples is present in the chemical state of Mn(IV).20
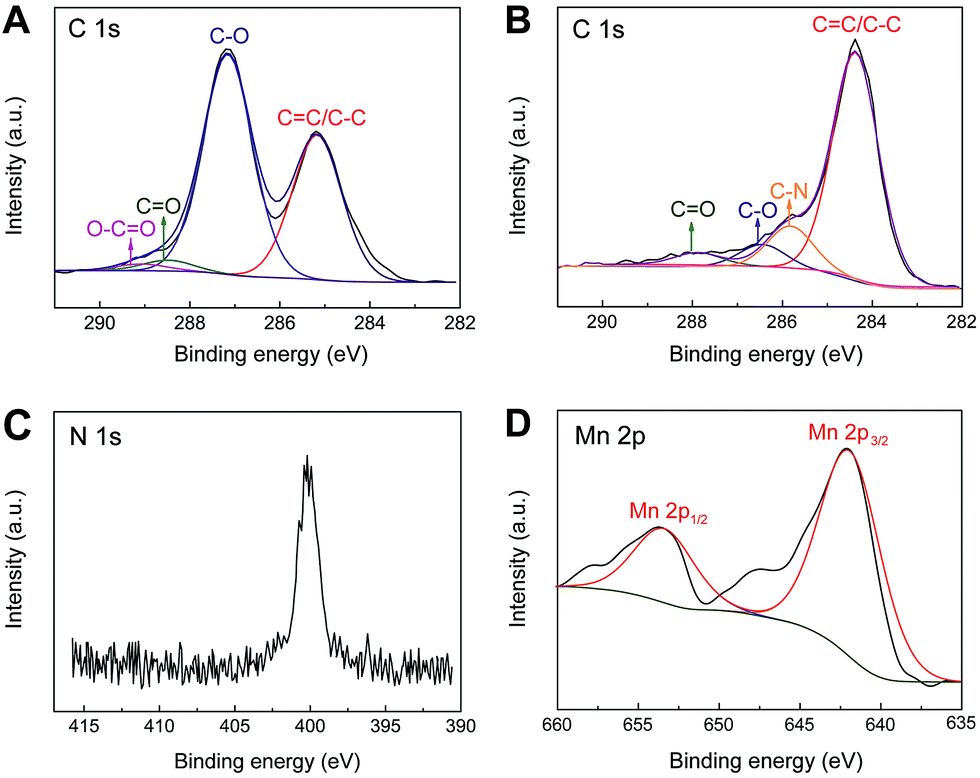 | ||
| Fig. 3 The XPS spectra of curve fit of C1s of (A) GO and (B) IL–CNT–rGO nanocomposite. (C) The XPS spectra of curve fit of N1s of IL–CNT–rGO nanocomposite. (D) The XPS spectra of curve fit of Mn2p of MnO2–rGO nanocomposite. | ||
Fig. 4 shows the X-ray diffraction (XRD) patterns of MnO2–rGO nanocomposite with controlled rGO and GO samples. The diffraction peak of GO is observed at about 2θ = 11.0°. After self-assembled into rGO gel under the solvothermal treatment, the characteristic peak of GO disappears, and a new broad peak at about 24.8° can be seen from rGO, indicating that GO has been completely reduced by thermal treatment. Comparing with rGO, the diffraction peak of MnO2–rGO nanocomposite at 24.8° broaden and decrease greatly, demonstrative that the decoration of MnO2 nanoparticles on graphene nanosheets can effectively prevent their restacking and aggregation during the one-step assembly process. In addition, two new peaks at 2θ values of about 36.5 and 66.0° are also observed, which are ascribed to the (211) and (002) crystal planes of α-MnO2, according to JCPDS card 44-0141.21 This indicates that the element Mn in MnO2–rGO nanocomposite exists in the formation of α-MnO2. According to X-ray spectroscopy (EDS) measurement, the content of MnO2 in MnO2–rGO nanocomposite has been calculated to be 72% (Fig. S2†). Furthermore, the mass ratio of MnO2 in MnO2–rGO sample calculated by thermal gravimetric analysis (TGA) is 78% (Fig. S3†), roughly consistent with that obtained by EDS analysis.
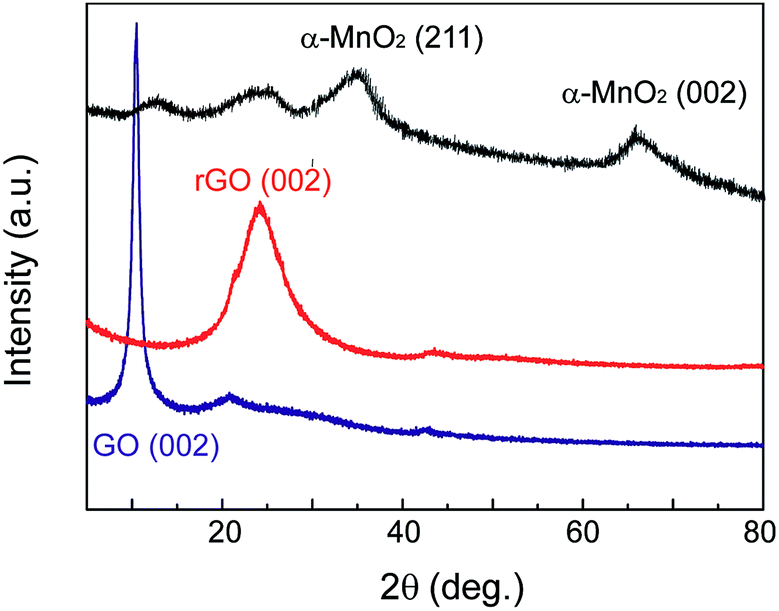 | ||
| Fig. 4 XRD patterns of GO, rGO and MnO2–rGO nanocomposite (from bottom to upper). | ||
Electrochemical characterization of different nanohybrid electrode materials
The electrochemical performances of IL–CNT–rGO nano-composite have been evaluated by cyclic voltammetry (CV) and galvanostatic (GV) charge/discharge in aqueous 1.0 M Na2SO4 electrolyte. A three-electrode system was adopted with the freestanding IL–CNT–rGO gel as the working electrode, and platinum wire and Ag/AgCl electrode as the counter and reference electrodes, respectively. The CV curves of IL–CNT–rGO electrode display a standard rectangular shape without any distinct redox peaks (Fig. 5A). The specific area of IL–CNT–rGO electrode has been calculated to be 1.87 V A g−1, which is much larger than that of 3D porous rGO (1.43 V A g−1), CNT–rGO (1.27 V A g−1) and IL–rGO (0.88 V A g−1) electrodes, as shown in Fig. 5B.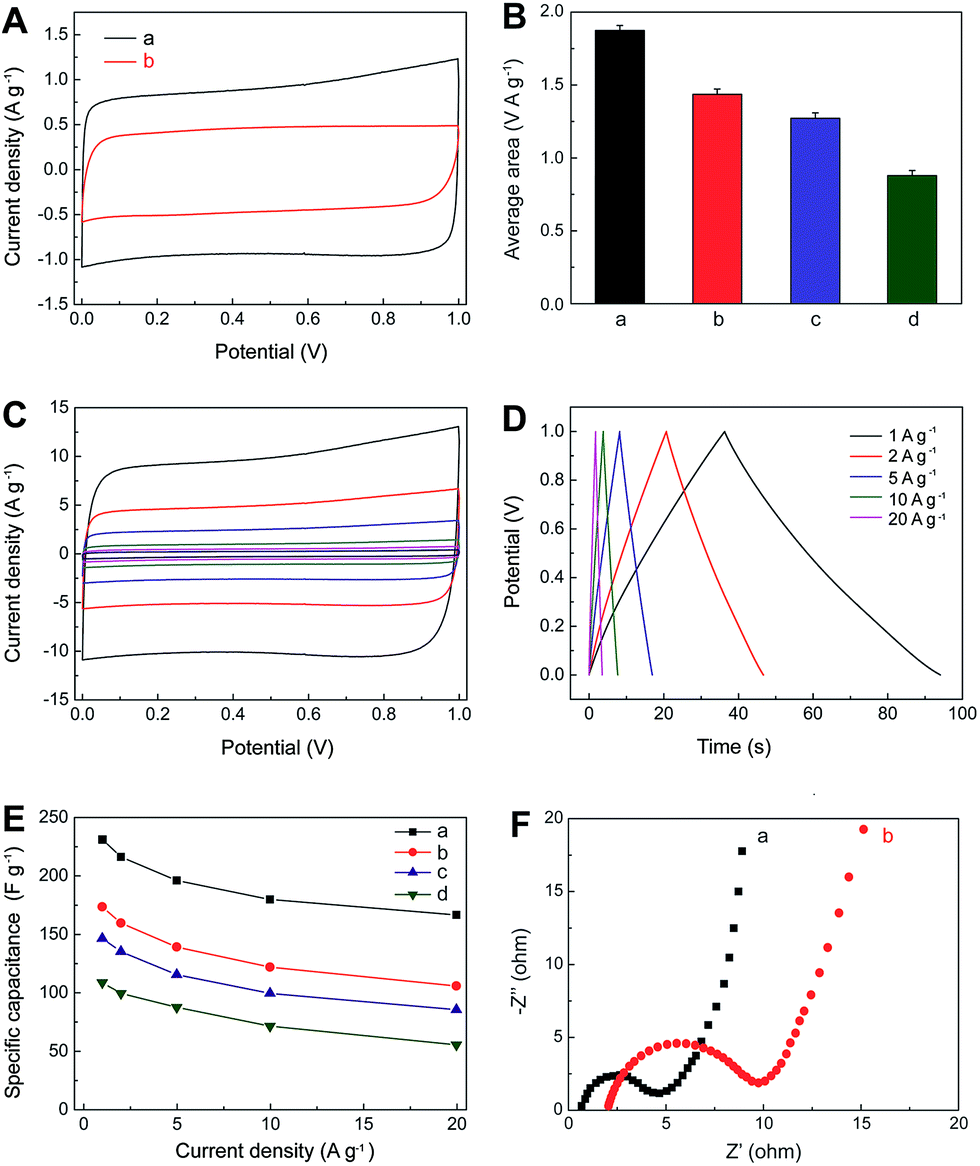 | ||
| Fig. 5 (A) CV curves for IL–CNT–rGO (a) and rGO (b) electrodes at the scan rate of 5 mV s−1 in 1.0 M aqueous Na2SO4 electrolyte. (B) Histograms of average area of CV curves for IL–CNT–rGO (a), CNT–rGO (b), IL–rGO (c) and rGO (d) electrodes at the same scan rate of 5 mV s−1. (C) CV curves for IL–CNT–rGO at different scan rates (5, 10, 20, 50, 100 and 200 mV s−1 from inner to outer) in 1.0 M aqueous Na2SO4 electrolyte. (D) GV charge/discharge curves of IL–CNT–rGO at different current densities (1–20 A g−1). (E) The capacitance retention of IL–CNT–rGO (a), CNT–rGO (b), rGO–IL (c) and rGO (d) as a function of discharge current densities derived from GV discharging curves. (F) EIS curves of IL–CNT–rGO (a) and rGO (b) electrodes. | ||
Rate dependent CV curves of IL–CNT–rGO electrode are carried out at different scan rates in the potential range of 0–1 V. The CV curves show no obvious deviation from rectangular shape for a wide scan range from 5 to 200 mV s−1 (Fig. 5C), indicating a low contact resistance in the SC. From GV charge/discharge curves in Fig. 5D, it can be seen that all the curves are highly linear and symmetrical at various current densities from 1 to 20 A g−1. This implies that the IL–CNT–rGO electrode has exceptional electrochemical reversibility and charge/discharge properties. Moreover, the IR drops on all curves are similar and not obvious, even at 20 A g−1, which indicate the low overall resistance and excellent capacitive properties of the IL–CNT–rGO nanohybrid electrode material.
The relationship curves between specific capacitance (Csc) and current density of IL–CNT–rGO with controlled CNT–rGO, IL–rGO and rGO electrodes are plotted in Fig. 5E. The Csc values range from 231.2 to 166.5 F g−1 as the scan rates increase from 1 to 20 A g−1, maintaining 72.0% of its specific capacitance at a high rate, which is much higher than that of CNT–rGO (60.9%), IL–rGO (58.2%) and pristine rGO (50.9%), demonstrative of a superior rate capability by synergistic contribution of CNT and IL in IL–CNT–rGO nanocomposite. The remarkable improved electrochemical supercapacitive activity and cycle stability of IL–CNT–rGO nanocomposite can be recognized as: (i) the graphene-based gel materials possess well-defined and interconnected 3D nanoporous network driven by π–π stacking interactions of graphene nanosheets, which exhibit multi-dimensional conductivity and large surface area. (ii) Due to the “spacer” of CNTs and IL in this 3D hierarchical IL–CNT–rGO structure, graphene sheets could be kept completely from restacking, which further increase the surface area of IL–CNT–rGO and provide more conductive channels to facilitate the access of the aqueous electrolyte ions onto the graphene surface. (iii) The IL–CNT–rGO nanocomposite is rich of the hydrophilic functional groups associated with BF4− anion on IL, which enhances the hydrophilic ability and also facilitates the access of solvated and charged ions.
Fig. 5F displays the electrochemical impedance spectra (EIS) of IL–CNT–rGO and rGO electrodes, both of which exhibit small semicircle over the high frequency range, followed by quite straight line in the low frequency region. The interfacial charge-transfer resistance of IL–CNT–rGO is calculated to be ∼4.6 Ω from the semicircle of the Nyquist plot, which is smaller than that of rGO electrode (8.7 Ω). These indicate a lowered diffusion resistance by the shortened proton diffusion path and an ideally polarizable capacitance of IL–CNT–rGO nanocomposite.
The electrochemical performance of MnO2–rGO nanocomposite has also been evaluated by CV and GV charge/discharge using the MnO2–rGO as the working electrode. As shown in Fig. 6A, the CV curve of MnO2–rGO exhibits a characteristic pseudocapacitive behavior. The leveled current separation between leveled anodic and cathodic currents for MnO2–rGO is much larger than that of rGO, which is due to the pseudocapacitive contribution of MnO2 along with the double layer contribution of graphene in MnO2–rGO nanocomposite. Fig. 6B shows the rate-dependent CVs at different scan rates, which do not lose their quasi-rectangular shapes even at high scan rate. The charging curves are symmetrical with their corresponding discharge counterparts, as well as their good linear voltage–time profiles (Fig. 6C), proving an ideal pseudo-capacitive feature with fast charge/discharge process. The Csc of MnO2–rGO electrode at the discharge current density of 1 A g−1 is calculated to be 256 F g−1, which is almost 2.5 times larger than that of the pristine rGO (96 F g−1) at the same discharge current density. With the increase of current density from 1 to 20 A g−1, the Csc of MnO2–rGO electrode decreases from 256 to 164 F g−1, the capacitance retention ratio is calculated to be 64.1% (Fig. 6D).
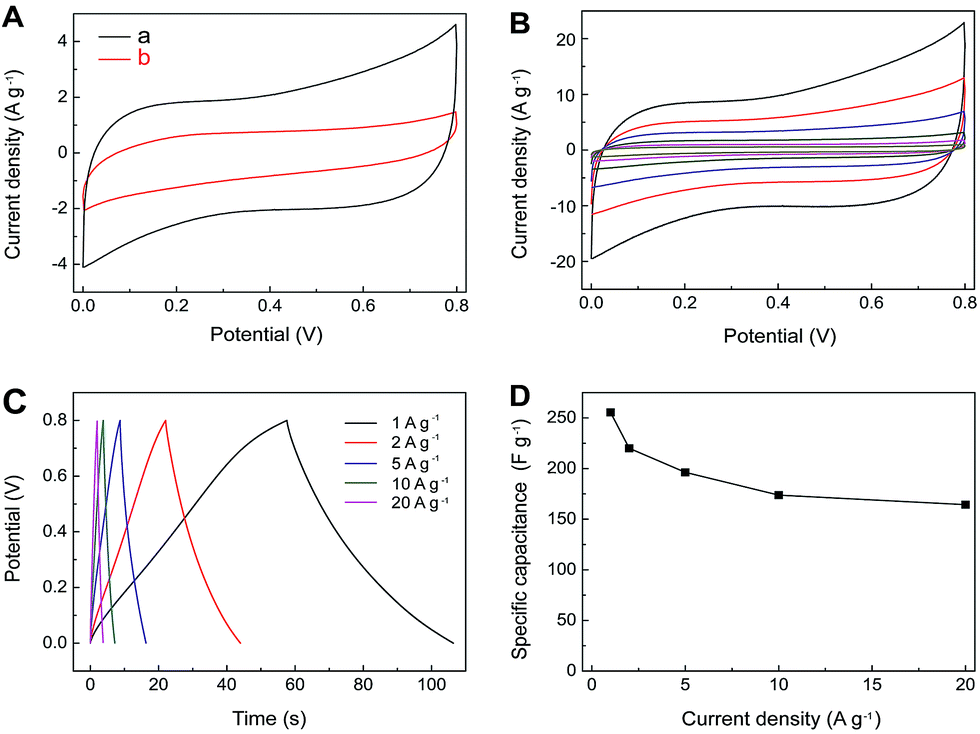 | ||
| Fig. 6 (A) CV curves for MnO2–rGO (a) and rGO (b) electrodes at the scan rate of 5 mV s−1 in 1.0 M aqueous Na2SO4 electrolyte. (B) CV curves for MnO2–rGO electrode at different scan rates (5, 10, 20, 50, 100 and 200 mV s−1 from inner to outer) in 1.0 M aqueous Na2SO4 electrolyte. (C) GV charge/discharge curves of MnO2–rGO electrode at different current densities (1–20 A g−1). (D) The capacitance retention of MnO2–rGO electrode as a function of discharge current densities derived from GV discharging curves. | ||
Electrochemical performance asymmetric hybrid SCs
Considering the fast ion-transport property of the electric double-layer storage for IL–CNT–rGO and the high pseudocapacitance of MnO2–rGO electrodes, an asymmetric SC has been fabricated using IL–CNT–rGO and MnO2–rGO as the negative and positive electrodes, respectively. The mass ratio of negative electrode to positive electrode is fixed close to 1:1, which is based on charge balance theory and their respective specific capacitances. The CV curve of IL–CNT–rGO electrode has a rectangular shape in the potential window from 0 to 1 V, while the CV curve of MnO2–rGO electrode shows pseudocapacitive behavior between 0 and 0.8 V (Fig. 7A). The total cell voltage of the asymmetric SC of IL–CNT–rGO//MnO2–rGO can be extended up to 1.8 V in 1.0 M aqueous Na2SO4 solution. Moreover, as shown in Fig. 7B, the CV curves of the optimized asymmetric SC exhibit rectangular-like shapes, with a cell voltage up to 1.8 V, and retain its original shape without obvious distortion with increasing potential scan rates even at a scan rate of 200 mV s−1. Fig. 7C shows the GV charge/discharge curves of the as-prepared asymmetric SC, which demonstrates an ideal capacitive behavior and a desirable fast charge/discharge property for SC device. From the slope of the discharge curve, the specific capacitance (Ct) of the IL–CNT–rGO//MnO2–rGO is calculated to be 57 F g−1 at a current density of 1 A g−1 (Fig. 7D). The asymmetric SC show a low capacitance loss about 10% after 10000 cycles (Fig. 7E), revealing its outstanding electrochemical stability. This can be attributed to the unique structure of IL–CNT–rGO gel and MnO2–rGO gel. The IL–CNT–rGO gel demonstrates a unique “skeleton/skin” structure with CNTs as the skeleton and graphene nanosheets as the skin. The “spacer” CNTs not only prevent the graphene nanosheets from restacking, but also enhance the mechanical strength of the as-obtained graphene gel. The MnO2–rGO gel exhibits a distinct “plum pudding” structure, where the MnO2 nanoparticles that decorated on 3D graphene gel are tightly wrapped by graphene nanosheets. This unique structure can help stabilize the MnO2 nanoparticles mechanically and avoid them loss from electrode surfaces due to Mn dissolution into electrolyte solutions. Therefore, the resultant asymmetric supercapacitor of IL–CNT–rGO gel//MnO2–rGO gel achieves high energy as well as good cycling stability and affordability.
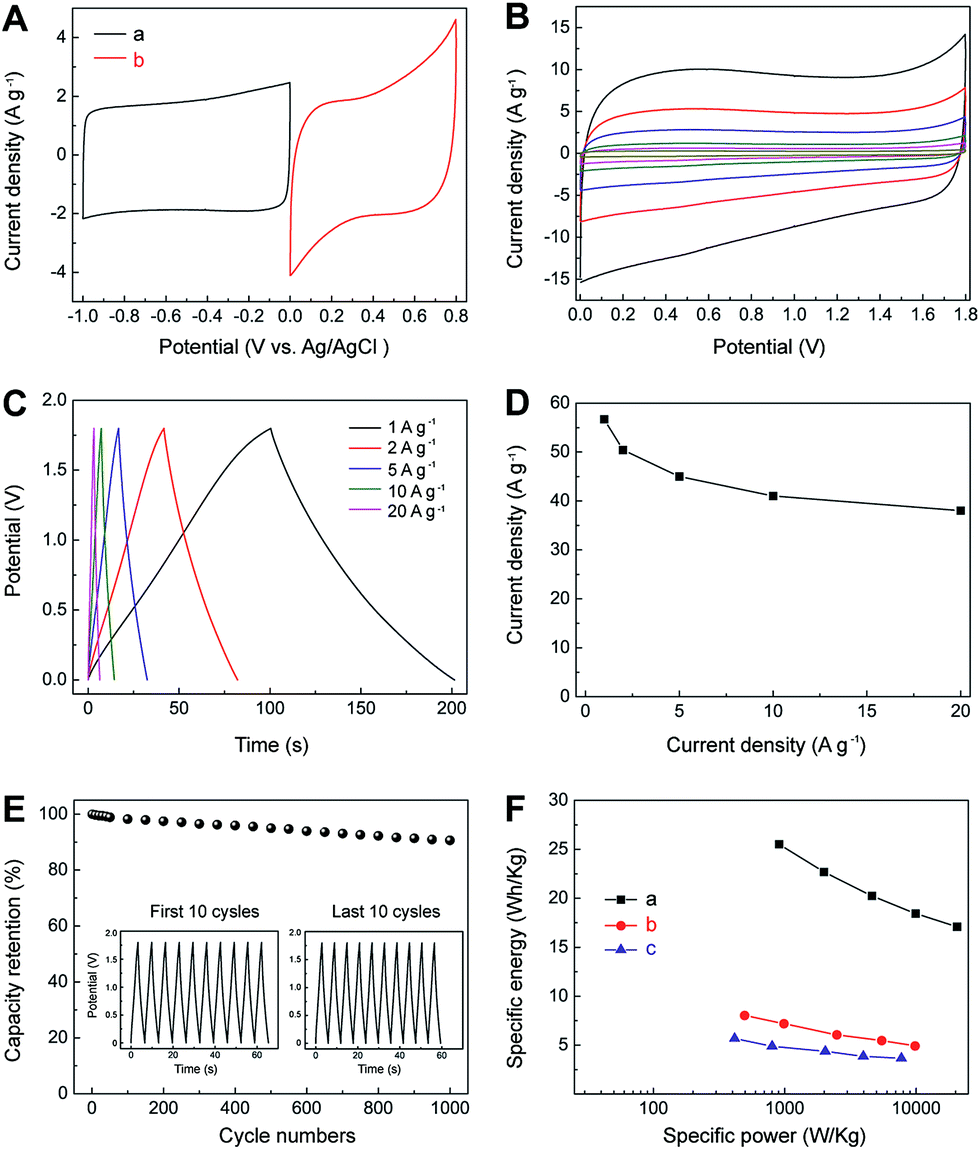 | ||
| Fig. 7 (A) Comparative CV curves of IL–CNT–rGO (a) and MnO2–rGO (b) in a three-electrode system in 1.0 M aqueous Na2SO4 solution at a scan rate of 5 mV s−1. (B) CV curves for IL–CNT–rGO//MnO2–rGO at different scan rates (5, 10, 20, 50, 100 and 200 mV s−1 from inner to outer) in 1.0 M aqueous Na2SO4 electrolyte. (C) GV charge/discharge curves of IL–CNT–rGO//MnO2–rGO at different current densities (1–20 A g−1). (D) The specific capacitance of IL–CNT–rGO//MnO2–rGO as a function of discharge current densities derived from GV discharging curves. (E) Charge/discharge cycling test at the current density of 20 A g−1. Inset shows the GV charge/discharge cyclic curves of the first and last 10 cycles. (F) Ragone plots of IL–CNT–rGO//MnO2–rGO (a), IL–CNT– rGO//IL–CNT–rGO (b) and MnO2–rGO//MnO2–rGO (c). | ||
Ragone plots, depicting the relationship of power densities and energy densities, are further used to evaluate the performance of the IL–CNT–rGO//MnO2–rGO asymmetric SC, IL–CNT–rGO//IL–CNT–rGO and MnO2–rGO//MnO2–rGO symmetric SCs. As shown in Fig. 7F, it can be seen that both power densities and energy densities of asymmetric SC are much higher than that of symmetric supercapacitors at the same current density. The energy density of the asymmetric SC of IL–CNT–rGO//MnO2–rGO is 26.5 W h kg−1 at a current density of 1 A g−1, which is about 3 or 5 folds than that of symmetric SCs of IL–CNT–rGO//IL–CNT–rGO (8.0 W h kg−1) and MnO2–rGO//MnO2–rGO (5.7 W h kg−1). More importantly, the asymmetric SC could provide a high energy density without sacrificing power density. When the power density increases to 20.5 kW kg−1, the energy density of IL–CNT–rGO//MnO2–rGO still retains 17.1 W h kg−1 at current density of 20 A g−1. It is found that the maximum energy density obtained for our asymmetric SC at a current density of 1 A g−1 is comparable or higher than those of MnO2 and/or carbon nanomaterials based asymmetric SCs with aqueous electrolyte solutions, such as graphene//graphene–MnO2 (21.27 W h kg−1 at a current density of 0.223 A g−1),22 activated carbon//NaMnO2 (19.5 W h kg−1 at a current density of 0.04 A g−1),23 activated carbon//MnO2 (17.3 W h kg−1 at a current density of 0.55 A g−1),24 graphene–patched CNT–MnO2 nanocomposite papers (24.8 W h kg−1),25 hierarchically structured Ni3S2/carbon nanotube composites (energy density of 19.8 W h kg−1 at a power density of 798 W kg−1),26 NiW-70//AC (15.1 W h kg−1 at 4.8 kW kg−1),27 NixCo1xLDH–ZTO//AC (9.7 W h kg−1 at 5.82 kW kg−1),28 NaMnO2//AC (13.2 W h kg−1 at 1.0 kW kg−1),29 MnO2 nanorods//AC (17 W h kg−1 at 2.0 kW kg−1),30 LiTi2(PO4)3//AC (15 W h kg−1 at 1.0 kW kg−1),31 MnO2//mesoporous carbon nanotubes ASC (10.4 W h kg−1 at 2 kW kg−1),32 graphene–MnO2//activated carbon nanofiber ASC (8.2 W h kg−1 at 16.5 kW kg−1),33 nanoarchitectured polyaniline/graphene–carbon nanotube (20.5 W h kg−1),34 Ni(OH)2/graphene//porous graphene ASC (13.5 W h kg−1 at 15.2 kW kg−1),35 MnO2//graphene hydrogel ASC (14.9 W h kg−1 at 10.0 kW kg−1),36 and graphene–MnO2//CNT textiles (12.5 W h kg−1).37
Conclusion
In summary, we developed a facile and green hydrothermal approach to fabricate two type of 3D porous graphene based materials, i.e., IL–CNT–rGO gel and MnO2–rGO gel, and explored their practical applications as freestanding and binder-free electrodes in asymmetric SC. Owing to the synergistic effects of different components in both IL–CNT–rGO and MnO2–rGO nanocomposites and the merits of one-step hydrothermal synthesis strategy, the as-obtained asymmetric SC of IL–CNT–rGO//MnO2–rGO exhibits good capacitive properties in terms of high specific capacitance, excellent rate capability, wide cell voltage, acceptable energy density and good cycling stability. We envision that our strategy for designing high-performance 3D rGO-based nanohybrid gel will offer new insights on designing functional nanomaterials toward lightweight and portable SCs, batteries, fuel cells, solar cell and other advanced energy related devices.Experimental
Synthesis of IL–CNT–rGO gel
GO was synthesized from graphite powder with modified Hummer's method reported elsewhere.38 The obtained GO was washed with distilled water and dried under vacuum. Then CNT and GO (dry powder, mass ratio 2:1) and IL (i.e., 1-butyl-3-methylimidazolium tetrafluoroborate, BMIMBF4) were mixed and ground in an agate mortar to form a uniform black gel (IL–CNT–GO gel), the ratio of CNT–GO nanocomposite to IL was 1 mg to 50 μL. The IL–CNT–GO suspension was fabricated by transferring aforementioned IL–CNT–GO gel into appropriate amount of N,N-dimethyl-formamide (DMF), ultrasonic agitating for a few minutes to give a black suspension, the ratio of CNT–GO to DMF was 9 mg to 1 mL. The suspension was then transferred into a Teflonlined stainless steel autoclave (20 mL) and heated 200 °C for 5 h. After reaction, the reactors were cooled to room temperature naturally. The as-obtained nanocomposite was denoted as CNT–rGO–IL gel. CNT–rGO gel and IL–rGO gel were prepared under the similar procedure. The as-prepared gel cylinders were washed carefully and throughly with water, and freeze-dried under vaccum.
Synthesis of MnO2–graphene gel
For the synthesis of MnO2–graphene gel, GO (30 mg) was dispersed in 10 mL DMF by sonication to form a homogenous suspension. Mn(CH3COO)2·4H2O (75 mg) was also dissolved in 1 mL of DMF, and then added into the obtained GO suspension. The resultant mixture was transferred into a Teflonlined stainless steel autoclave (20 mL) and heated 200 °C for 5 h. After this reaction, the reactors were cooled to room temperature naturally. The resultant MnO2–rGO gel cylinder was washed carefully and throughly with water, and freeze-dried under vaccum. The rGO gel was prepared under the same procedure.Characterization
The microstructures of the materials were examined by field-emission scanning electron microscope (SEM) (JSM-6701F, JEOL) and transmission electron microscope (TEM) (JEOL JEM2010). For the preparation of TEM samples, the graphene-based gel materials were firstly dispersed in water under ultrasonic assistance to form a homogenous suspension, which were deposited on Cu grid and drying at the room temperature for TEM characterization. SEM was equipped to perform elemental analysis by energy dispersive X-ray spectroscopy (EDS). X-ray diffraction (XRD) patterns were collected with a Bruker AXS D8 X-ray diffractometer equipped with monochromatized Cu Kα radiation (λ = 1.54056 Å, 40 kV, and 20 mA). X-ray photoelectron spectroscopy (XPS) was performed on a Kratos-Axis spectrometer with monochromatic Al Kα (hν = 1486.71 eV) X-ray radiation (15 kV and 10 mA) and a hemispherical electron energy analyzer. All XPS spectra were corrected according to the C1s line at 284.6 eV. Curve fitting and background subtraction were accomplished using Casa XPS software. The specific surface areas and pore size distributions of the electrode materials were calculated by Brunauer–Emmett–Teller (BET) and Barrett–Joyner–Halenda (BJH) methods, respectively. Thermogravimetric Analysis (TGA) was acquired with TA Instruments TGA/DSC. Cyclic voltammetry (CV), galvanostatic (GV) charge/discharge, and electrochemical impedance testes of SCs were carried out on a CHI 660D electrochemical workstation.Electrochemical test
IL–CNT–rGO gel and MnO2–rGO gel were cut into pieces and used as freestanding and binder-free electrodes for electrochemical characterization. The electrochemical tests of individual electrode were performed in a three-electrode cell, in which platinum foil and Ag/AgCl (4 M KCl) electrodes were used as counter and reference electrodes, respectively. To construct the asymmetric SC, the loading mass ratio of active materials (i.e., IL–CNT–rGO gel and MnO2–rGO gel) was estimated to be 1:1 from the specific capacitance calculated from their CV curves in a three-electrode cell. The asymmetric SC was built with a glassy fibrous separator and performed in a two-electrode cell in 1.0 M aqueous Na2SO4 electrolyte solution without removal of oxygen from the solution.
Calculation of specific capacitance, energy density and power density
The specific capacitance (Ct) of a SC cell is calculated using the equations Ct = IΔt/mΔV, where I is the constant discharge current, Δt is the discharging time, m is the total mass of active materials in both electrodes, and ΔV is the voltage drop upon discharging (excluding IR drop). In symmetric SCs, the specific capacitance (Csc) of the electrode is calculated following the equations Csc = 4Ct. The energy density (E) and power density (P) of a SC cell in the Ragone plots are calculated using the equations of E = CtΔV2/2 and P = E/Δt, respectively.39Acknowledgements
This work is supported by the National Natural Science Foundation of China (Grant 50773059) and the program of Nanyang Assistant Professorship of Nanyang Technological University, Singapore.Notes and references
- A. Manthiram, A. V. Murugan, A. Sarkar and T. Muraliganth, Energy Environ. Sci., 2008, 1, 621 Search PubMed; M. Winter and R. J. Brodd, Chem. Rev., 2004, 104, 4245 CrossRef CAS; J. R. Miller and P. Simon, Science, 2008, 321, 651 CrossRef PubMed.
- B. E. Conway, Electrochemical Supercapacitors: Scientific Fundamentals and Technological Applications, Plenum, New York, 1999 CrossRef CAS PubMed; P. Simon and Y. Gogotsi, Nat. Mater., 2008, 7, 845 CrossRef CAS PubMed.
- Y. Cheng, H. Zhang, S. Lu, C. V. Varanasi and J. Liu, Nanoscale, 2013, 5, 1067 RSC.
- M. Zhi, C. Xiang, J. Li, M. Li and N. Wu, Nanoscale, 2013, 5, 72 RSC.
- Z. Fan, J. Yan, T. Wei, L. Zhi, G. Ning, T. Li and F. Wei, Adv. Funct. Mater., 2011, 21, 2366 CrossRef CAS.
- Z. S. Wu, W. Ren, D. W. Wang, F. Li, B. Liu and H. M. Cheng, ACS Nano, 2010, 4, 5835 CrossRef CAS PubMed.
- M. Armand, F. Endres, D. R. MacFarlane, H. Ohno and B. Scrosati, Nat. Mater., 2009, 8, 621 CrossRef CAS PubMed.
- J. Tao, N. Liu, L. Li, J. Su and Y. Gao, Nanoscale, 2014, 6, 2922 RSC.
- B. G. Choi, S. J. Chang, H. W. Kang, C. P. Park, H. J. Kim, W. H. Hong, S. G. Lee and Y. S. Huh, Nanoscale, 2012, 4, 4983 RSC.
- Q. Lu, J. G. Chen and J. Q. Xiao, Angew. Chem., Int. Ed., 2013, 52, 1882 CrossRef CAS PubMed.
- A. K. Geim and K. S. Novoselov, Nat. Mater., 2007, 6, 183 CrossRef CAS PubMed; S. Stankovich, D. A. Dikin, G. H. B. Dommett, K. M. Kohlhaas, E. J. Zimney, E. A. Stach, R. D. Piner, S. T. Nguyen and R. S. Ruoff, Nature, 2006, 442, 282 CrossRef PubMed.
- J. Li, C. Y. Liu and Y. Liu, J. Mater. Chem., 2012, 22, 8426 RSC; C. Y. Hou, Q. H. Zhang, Y. G. Li and H. Z. Wang, Carbon, 2012, 50, 1959 CrossRef CAS PubMed; H. P. Cong, X. C. Ren, P. Wang and S. H. Yu, ACS Nano, 2012, 3, 2693 CrossRef PubMed; Y. Xu, K. Sheng, C. Li and G. Shi, ACS Nano, 2010, 4, 4324 CrossRef PubMed; J. Wang, H. Jin, Y. He, D. Lin, A. Liu, S. Wang and J. Wang, Nanoscale, 2014, 6, 7204 RSC; T. Sun, Z. Zhang, J. Xiao, C. Chen, F. Xiao, S. Wang and Y. Liu, Sci. Rep., 2013, 3, 2527 Search PubMed; Z. Zhang, F. Xiao, Y. Guo, S. Wang and Y. Liu, ACS Appl. Mater. Interfaces, 2013, 5, 2227 Search PubMed; Z. Zhang, F. Xiao, L. Qian, J. Xiao, S. Wang and Y. Liu, Adv. Energy Mater., 2014, 1400064 Search PubMed.
- C. Li and G. Shi, Adv. Mater., 2014, 26, 3992 CrossRef CAS PubMed.
- Y. C. Si and E. T. Samulski, Chem. Mater., 2008, 20, 6792 CrossRef CAS; Y. Sun, Q. Wu and G. Shi, Energy Environ. Sci., 2011, 4, 1113 Search PubMed.
- N. Liu, F. Luo, H. Wu, Y. Liu, C. Zhang and J. Chen, Adv. Funct. Mater., 2008, 18, 1518 CrossRef CAS; H. Yang, C. Shan, F. Li, D. Han, Q. Zhang and L. Niu, Chem. Commun., 2009, 3880 RSC; A. A. J. Torriero, A. I. Siriwardana, A. M. Bond, I. M. Burgar, N. F. Dunlop, G. B. Deacon and D. R. MacFarlane, J. Phys. Chem. B, 2009, 113, 11222 CrossRef PubMed; M. J. A. Shiddiky and A. A. J. Torriero, Biosens. Bioelectron., 2011, 26, 1775 CrossRef PubMed; F. Xiao, F. Zhao, D. Mei, Z. Mo and B. Zeng, Biosens. Bioelectron., 2009, 24, 3481 CrossRef PubMed; F. Xiao, F. Q. Zhao, Y. F. Zhang, G. P. Guo and B. Z. Zeng, J. Phys. Chem. C, 2009, 113, 849 Search PubMed; B. G. Choi, Y. Suk Huh, W. H. Hong, H. J. Kim and H. S. Park, Nanoscale, 2012, 4, 5394 RSC.
- T. Y. Kim, H. W. Lee, M. Stoller, D. R. Dreyer, C. W. Bielawski, R. S. Ruoff and K. S. Suh, ACS Nano, 2011, 5, 436 CrossRef CAS PubMed.
- C. Zhu, J. Zhai and S. Dong, Nanoscale, 2014, 6, 10077 RSC; T. Fukushima, A. Kosaka, Y. Ishimura, T. Yamamoto, T. Takigawa, N. Ishii and T. Aida, Science, 2003, 300, 2072 CrossRef CAS PubMed.
- Y. Hou, Y. W. Cheng, T. Hobson and J. Liu, Nano Lett., 2010, 10, 2727 CrossRef CAS PubMed.
- F. Xiao, J. Song, H. Gao, X. Zan, R. Xu and H. Duan, ACS Nano, 2012, 6, 100 CrossRef CAS PubMed; H. Gao, F. Xiao, C. B. Ching and H. Duan, ACS Appl. Mater. Interfaces, 2011, 3, 3049 Search PubMed.
- L. Mao, K. Zhang, H. S. O. Chan and J. Wu, J. Mater. Chem., 2012, 22, 1845 RSC.
- J. Yan, Z. Fan, T. Wei, W. Qian, M. Zhang and F. Wei, Carbon, 2010, 48, 3825 CrossRef CAS PubMed.
- L. J. Deng, G. Zhu, J. F. Wang, L. P. Kang, Z. H. Liu, Z. P. Yang and Z. L. Wang, J. Power Sources, 2011, 196, 10782 CrossRef CAS PubMed.
- Q. T. Qu, Y. Shi, S. Tian, Y. H. Chen, Y. P. Wu and R. J. Holze, J. Power Sources, 2009, 194, 1222 CrossRef CAS PubMed.
- T. Cottineau, M. Toupin, T. Delahaye, T. Brousse and D. Belanger, Appl. Phys. A: Mater. Sci. Process., 2006, 82, 599 CrossRef CAS PubMed.
- Y. Jin, H. Chen, M. Chen, N. Liu and Q. Li, ACS Appl. Mater. Interfaces, 2013, 5, 3408 CAS.
- C. S. Dai, P. Y. Chien, J. Y. Lin, S. W. Chou, W. K. Wu, P. H. Li, K. Y. Wu and T. W. Lin, ACS Appl. Mater. Interfaces, 2013, 5, 12168 CAS.
- L. Niu, Z. Li, Y. Xu, J. Sun, W. Hong, X. Liu, J. Wang and S. Yang, ACS Appl. Mater. Interfaces, 2013, 5, 8044 CAS.
- W. Wang, A. Sumboja, M. Lin, J. Yan and P. S. Lee, Nanoscale, 2012, 4, 7266 RSC.
- Q. T. Qu, Y. Shi, S. Tian, Y. H. Chen, Y. P. Wu and R. Holze, J. Power Sources, 2009, 194, 1222–1225 CrossRef CAS PubMed.
- Q. Qu, P. Zhang, B. Wang, Y. Chen, S. Tian, Y. Wu and R. Holze, J. Phys. Chem. C, 2009, 113, 14020 CAS.
- J. Y. Luo and Y. Y. Xia, J. Power Sources, 2009, 186, 224 CrossRef CAS PubMed.
- H. Jiang, C. Z. Li, T. Sun and J. Ma, Nanoscale, 2012, 4, 807 RSC.
- Z. J. Fan, J. Yan, T. Wei, L. J. Zhi, G. Q. Ning, T. Y. Li and F. Wei, Adv. Funct. Mater., 2011, 21, 2366 CrossRef CAS.
- J. Shen, C. Yang, X. Li and G. Wang, ACS Appl. Mater. Interfaces, 2013, 5, 8467 CAS.
- L. Zhang, J. Zhi and F. Wei, Adv. Funct. Mater., 2012, 22, 2632 CrossRef.
- H. Gao, F. Xiao, C. B. Ching and H. Duan, ACS Appl. Mater. Interfaces, 2012, 4, 2801 CAS.
- G. H. Yu, L. B. Hu, M. Vosgueritchian, H. L. Wang, X. Xie, J. R. McDonough, X. Cui, Y. Cui and Z. N. Bao, Nano Lett., 2011, 11, 2905 CrossRef CAS PubMed.
- W. S. Hummers and R. E. Offeman, J. Am. Chem. Soc., 1958, 80, 1339 CrossRef CAS.
- M. D. Stoller and R. S. Ruoff, Energy Environ. Sci., 2010, 3, 1294 Search PubMed; T. Brousse, M. Toupin and D. J. Belanger, J. Electrochem. Soc., 2004, 151, A614 CrossRef CAS PubMed.
Footnote |
| † Electronic supplementary information (ESI) available: BET of IL–CNT–rGO gel and MnO2–rGO gel, and EDS spectrum and TGA of MnO2–rGO gel are presented. See DOI: 10.1039/c4ra16071a |
| This journal is © The Royal Society of Chemistry 2015 |